

CRISPR/Cas9 gene editing has become an increasingly popular solution for introducing permanent loss-of-function (LOF) mutations by generating double-strand breaks at targeted genomic loci, after which endogenous cellular machinery repairs the cleaved DNA. LOF stems from the introduction of frameshift mutations during this repair process. The adaptation of the CRISPR/Cas9 system expands the application of this technology, allowing CRISPR-mediated gene activation (CRISPRa) and repression (CRISPRi) without altering the DNA.
How CRISPRa and CRISPRi Work
In contrast to regular CRISPR/Cas9 gene editing, CRISPRa and CRISPRi employ a catalytically inactive form of Cas9 (dCas9). This Cas9 variant possesses point mutations in two amino acid residues, D10A and H840A, that deactivate the RuvC and HNH nuclease domains of Cas9, respectively. [1] Although dCas9 cannot cut DNA, it is still precisely recruited to the target DNA by the guide RNA (gRNA). Synthetic biologists have taken advantage of this property of dCas9 by constructing dCas9-based tools that expand the functionality of the CRISPR/Cas9 system.
dCas9 can be transformed into a transcriptional activator or repressor by fusing or otherwise recruiting the appropriate transcriptional effector domains to the inactive nuclease (Figure 1). Commonly used transcriptional activator domains include VP64, the p65 domain of NF-\(\kappa\)B, the Epstein Barr virus R transactivator (Rta), and the activator domain for heat shock factor 1 (HSF1). [2] The primary transcriptional repressor used in combination with dCas9 is the Krüppel associated box (KRAB) domain from KOX1, forming dCas9 KRAB. [3]
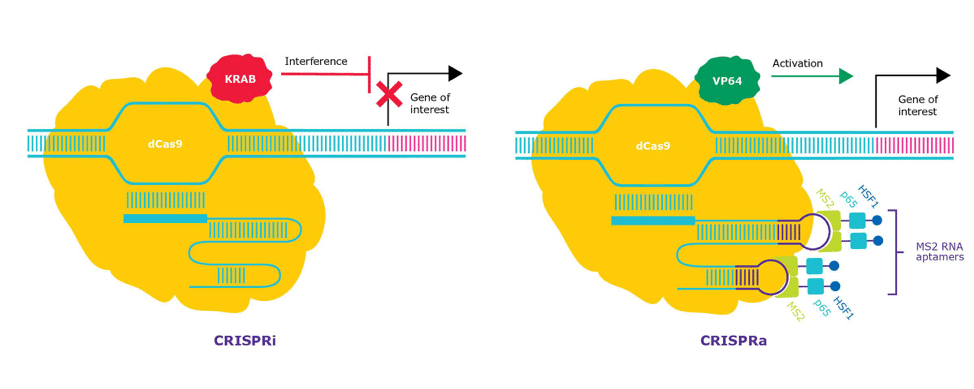
In the endogenous context, multiple transcription factors and cofactors work in synchrony to stimulate gene transcription. Indeed, CRISPRa tools that recruit multiple unique transcriptional activators to a promoter outperform those bearing a single transcriptional activator domain or redundant copies of the same effector. [4,5] Targeting multiple sites on the same promoter also increases gene activation with CRISPRa. [5] One of the most effective CRISPRa effectors is the CRISPR Synergistic Activation Mediator (SAM) complex, which recruits three unique transcriptional activator domains to the targeted gene promoter. [4, 5] In this system, one transcriptional activator VP64 (a multimeric form of VP16) is directly fused to dCas9. Protein-binding RNA aptamers engineered into the stem-loop regions of the gRNA recruit the other two transcriptional activator domains. [5] CRISPRi and CRISPRa complexes only need to be expressed alongside an appropriately designed gRNA to induce CRISPRi or CRISPRa in mammalian cells.
Considerations for Designing a CRISPRa or CRISPRi Experiment
How to design the gRNA
The design of gRNAs differs between CRISPRa or CRISPRi and CRISPR KO, where efficient gRNAs target early exons in protein-coding genes to prevent the expression of a truncated protein that may retain some function. A screen of all possible gRNAs surrounding the transcriptional start site (TSS) of 49 genes enabled the identification of the optimal targeting windows for both CRISPRi and CRISPRa. [6]
Consistent with its dual mechanism of action, CRISPRi using the dCas9-KRAB repressor was efficient for gRNAs falling in a window spanning from -50 to +300 base pairs (bp) from each TSS, with the best-performing gRNAs targeting the first 100 bp downstream of the TSS. [6] For CRISPRa, the window spanning -400 to -50 bp from an individual TSS was determined to be the optimal targeting region. [6] This window applies to all CRISPRa effectors, including the SAM system. [5]
Since the efficacy of a CRISPRa or CRISPRi gRNA is impacted by the proximity to the TSS of the targeted gene, meaning gRNA design for CRISPRa or CRISPRi is more complicated for poorly annotated genomes. The efficacy of gRNAs for CRISPRa or CRISPRi is reduced by:
- Long protospacer lengths (>21 bp).
- The presence of nucleotide homopolymers (e.g., AAAA or GGGG) in the protospacer region.
Also, the chromatin environment surrounding the targeted genomic region can limit the nuclease accessing the site. [7]
Identifying highly active gRNAs is vital for constructing genome-scale CRISPRi and CRISPRa libraries. Such libraries include 3-10 gRNAs per gene to ensure that screening hits are not because of inadequate gRNA efficacy. [8, 9] Pooled gRNA screens offer a robust and straightforward method for clarifying how various factors influence gRNA activity, as thousands of gRNAs can be tested with relatively minimal effort. [10]
Pooled gRNA screens also inform gRNA prediction algorithms. Observations, such as the effects of gRNA sequence, position, and accessibility, have enabled the iterative and comprehensive identification of highly active gRNAs. [11, 12] This has led to the development of highly active and compact (i.e., fewer gRNAs per gene) gRNA libraries for CRISPRi and CRISPRa. [9] Ultimately, compact CRISPR libraries reduce the number of cells needed to perform genome-scale screens.
The Sigma-Aldrich® suite of functional genomics solutions includes optimized, whole-genome gRNA libraries for CRISPRi and CRISPRa as well as custom gRNA design services for individual targets and large-scale libraries. Discover detailed protocols for CRISPRi and CRISPRa screening.
CRISPRa and CRISPRi Compared to Competing Technologies
The mechanisms of CRISPRi and CRISPRa differ from other gene perturbation technologies, such as RNA interference (RNAi) and CRISPR KO (Figure 2). These differences offer advantages and challenges that should be taken into consideration when selecting which technology to implement.
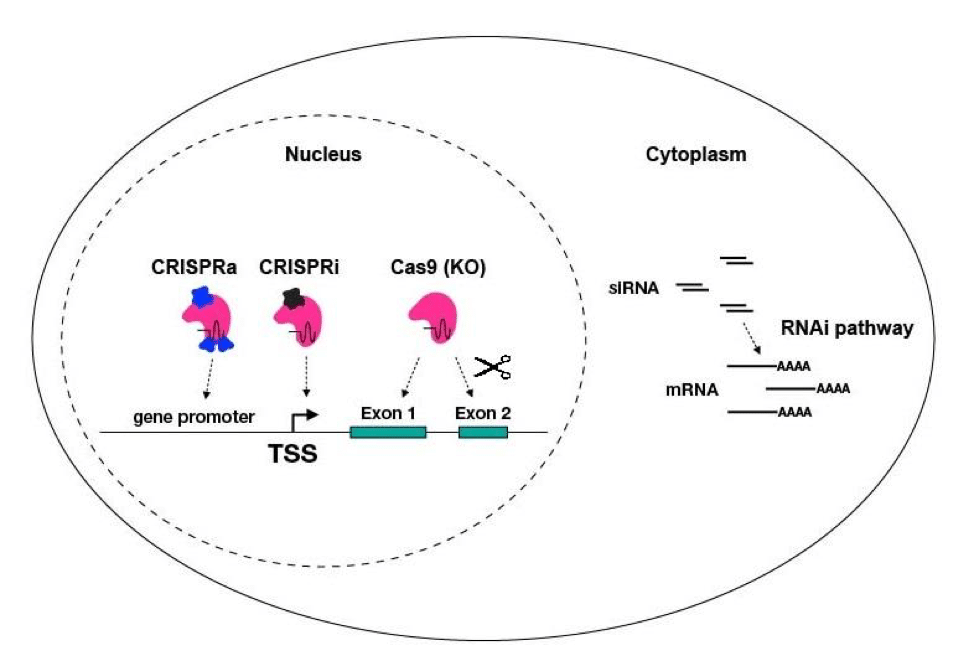
CRISPRi vs. RNAi
The mechanism of CRISPRi differs from other gene perturbation technologies, such as RNA interference (RNAi). RNAi inhibits gene expression by utilizing a conserved RNA processing pathway to degrade targeted mRNAs in the cytoplasm. The cytoplasmic location of the endogenous machinery co-opted by RNAi prevents it from efficiently targeting nuclear RNA, such as ncRNA. [13] RNAi can also induce off-target effects by displacing endogenous small RNAs (e.g., microRNAs) from RISC. [14] CRISPRi avoids these unwanted effects because all of the components are from exogenous sources. Finally, CRISPRi outperforms RNAi in large-scale screening applications by generating more robust phenotypes with fewer off-target effects. [6]
CRISPRi vs. CRISPR KO
Although CRISPR gene editing is a powerful method for generating LOF mutants, several disadvantages preclude its use in all LOF studies. The double-strand breaks caused by Cas9 can induce cytotoxicity and genomic instability, particularly in cancer cell lines. [15,16] Also, roughly one-third of indels will not cause the necessary frameshift to knock out targeted genes. Non-coding regions (e.g., long non-coding RNAs) can be difficult to target by Cas9 because they require edits at two sites to create a sufficiently large mutation to generate LOF. [17] CRISPR KO is also a suboptimal solution for investigating the effects of genes that are essential for cell survival beyond their identification in genome-wide screens as cells can tolerate a partial knockdown, but not a complete knockout, of such genes. [10] By not cutting DNA, CRISPRi circumvents the disadvantages of CRISPR KO, making it a superior choice for perturbation of non-coding genes or applications that call for a reversible or titratable solution.
CRISPRa vs. ORF Overexpression
CRISPRa offers a few advantages over existing gain-of-function (GOF) techniques such as open reading frame (ORF) overexpression. Overexpression of ORFs commonly utilizes strong viral promoters (e.g., CMV) that result in supraphysiological expression levels. Because CRISPRa is targeted to endogenous gene promoters, supraphysiological transcription is challenging to achieve, making CRISPRa better suited to applications that favor the physiological or near-physiological expression of targeted genes. [18] This also means CRISPRa is more likely to upregulate the most relevant splice variants of the targeted gene, particularly in cases when they are unknown. [10] Finally, genome-scale ORF libraries are more difficult to synthesize than equally-scaled CRISPRa libraries.
CRISPRi and CRISPRa add to the existing suite of genome engineering technologies by offering complementary insights into complex biological processes. Unlike CRISPR KO, which provides binary and permanent outcomes, CRISPRa and CRISPRi can be used to reversibly titrate gene expression levels within a broad, dynamic range (up to and exceeding 1,000-fold). [2] Also, large-scale LOF and GOF screens can identify unique, yet functionally related, hits under otherwise identical screening parameters. The identification of SPI1 (by CRISPRa) and GATA1 (by CRISPRi) as regulators of cell growth in the K562 human myeloid leukemia cell line highlights the usefulness of this parallel use of CRISPRi and CRISPRa. [6] Both manipulations increased cell growth in K562 cells, confirming SPI1’s known inhibitory effects on GATA1 activity. [19] Therefore, while individual CRISPR screens are independently powerful tools, when used in combination, CRISPRi, CRISPRa, and traditional CRISPR KO screens offer robust validation and strengthen findings, as well as provide a more detailed understanding of gene pathways.
How to Perform a CRISPRa or CRISPRi Experiment
A typical CRISPRa or CRISPRi experiment involves three necessary steps:
- Ideally, the generation of a stable cell line that expresses the modified Cas9 with the appropriate CRISPRa or CRISPRi constructs.
- Delivery of the gRNA.
- Analysis of the results.
Below is a brief overview of these steps; a more thorough protocol is available here.
Generation of a Helper Cell Line
Transient transfection of all components is sufficient for cell lines that are efficiently transfected (e.g., HEK293 cells), while lentiviral transduction provides more consistent and robust results across a variety of cell lines. For large-scale screening applications, creating a stable “helper” cell line that expresses dCas9-KRAB or the CRISPR SAM complex at the outset permits rapid validation at the end of the screening process. Sigma-Aldrich® lentiviral vectors are available for helper cell line generation that are optimized to improve functional viral titers.
gRNA Delivery
Lentiviral transduction of gRNA followed by antibiotic selection ensures consistent and robust activation or repression for both small and large-scale applications. Lentiviral delivery is essential for pooled CRISPR screens because genomically-integrated gRNA sequences serve as barcodes for later deconvolution. As such, pooled CRISPR libraries should be delivered at a low (<0.7) multiplicity of infection to ensure each cell receives only a single CRISPR copy. [8]
Analysis
For small-scale applications, analysis involves measuring changes in mRNA and protein levels using quantitative polymerase chain reaction (qPCR) and western blotting, respectively, as well as the functional consequences of those changes. Pooled CRISPR screens use next-generation sequencing to analyze changes in the representation of lentivirally transduced gRNAs following a selection process.
Applications of CRISPRa and CRISPRi
CRISPRa and CRISPRi are particularly amenable to genome- and subgenome-scale screening applications, including identifying drug targets [20] and cellular factors that mediate drug resistance, [5] due to the relative ease with which gRNA libraries can be synthesized, cloned, and packaged in lentivirus. [6, 5] Arrayed screening is also possible with CRISPRa and CRISPRi. Two SigmaAldrich® webinars provide more insight into CRISPR screening including the benefits of a whole-genome gain-of-function screening and the differences between CRISPRa, CRISPRi and CRISPR KO screens.
Although pooled CRISPR screening has many advantages over arrayed screening, it is typically limited to studying crude phenotypes such as cell growth and viability. By contrast, virtually any type of analysis can be done in an arrayed screen because CRISPR perturbations occur in individual wells of microtiter plates. The advent of single-cell RNA sequencing, however, provided a high-content transcriptomic analysis that could be integrated into pooled CRISPR screen workflows. This is because each CRISPR perturbation, including its transcriptomic and phenotypic effects, is confined to an individual cell. [21] Thus, as long as individual cells can be separated, CRISPR perturbations can be linked to a transcriptomic readout as well as a phenotypic outcome. CRISPR single-cell screening in partnership with 10x Genomics is now available from the Sigma-Aldrich® portfolio.
CRISPRa can be used to study non-coding genes [6] or individual transcript variants of a gene, [22] although well-annotated genomes are crucial for these applications. CRISPRi is also able to distinguish between different transcript variants, unlike CRISPR KO and RNAi, as long as the TSS for each variant is mapped.
CRISPRa and CRISPRi Tools Summarized
CRISPRa and CRISPRi are potent tools for modulating gene expression in a reversible and titratable manner. These technologies use dCas9 to act as synthetic transcription factors by recruiting endogenous transcription activator and repressor complexes to gene promoters and enhancers, resulting in up- or downregulated gene transcription. CRISPRi achieves LOF phenotypes without the limitations of RNAi and CRISPR KO, and CRISPRa provides a scalable solution for large-scale overexpression screens. Although CRISPRa and CRISPRi usually rely on delivery by lentivirus, these technologies offer new possibilities for genome engineering. CRISPRi and CRISPRa can be used individually to target areas of the genome that are inaccessible by other gene perturbation technologies (e.g., non-coding regions) or in conjunction to uncover gene-regulatory networks underlying discrete phenotypes. Finally, pooled CRISPRi and CRISPRa screening can be paired with single-cell RNA-seq to enable high-dimensional characterization of CRISPR perturbations.
More CRISPR Resources
CRISPR Articles
- CRISPR Gene Editing: Considerations and Getting Started
- How to Validate a CRISPR Experiment
- How to Understand CRISPR Formats and Their Applications
- Level Up Your Drug Screening With CRISPR
CRISPR Webinars
- Engineering Vero Cell Lines Using CRISPR to Increase Production of Viral Vaccines
- Efficient Generation of Gene-edited Mouse Models and Cell Lines Using Synthetic sgRNA
- CRISPR Screening: from design and discovery to validation
- A guide to leveraging single cell CRISPR screens for deeper biological insights
CRISPR eBook
CRISPR Gene Editing 101 (eBook)
CRISPR Tools
And you should also consider the following SigmaAldrich® tools:
- Sigma-Aldrich® CRISPR
- Sigma-Aldrich® Advanced Genomics
- Advanced Genomics Resource Center
- CRISPR Essentials
- Additional CRISPR Webinars
Discover more about CRISPR in the Bitesize Bio CRISPR Research Hub.
References
- Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-21. DOI: 10.1126/science.1225829.
- La Russa MF, et al. The New State of the Art: Cas9 for Gene Activation and Repression. Mol Cell Biol. 2015;35(22):3800-9. DOI: 10.1128/MCB.00512-15.
- Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442-51. DOI: 10.1016/j.cell.2013.06.044.
- Yang J, et al. Genome-Scale CRISPRa Screen Identifies Novel Factors for Cellular Reprogramming. Stem Cell Reports. 2019;12(4):757-71. DOI: 10.1016/j.stemcr.2019.02.010.
- Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583-8. DOI: 10.1038/nature14136.
- Gilbert LA, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159(3):647-61. DOI: 10.1016/j.cell.2014.09.029.
- Horlbeck MA, et al. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife. 2016;5. DOI: 10.7554/eLife.12677.
- Joung J, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12(4):828-63. DOI: 10.1038/nprot.2017.016.
- Horlbeck MA, et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016;5. DOI: 10.7554/eLife.19760.
- Doench JG. Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Genet. 2018;19(2):67-80. DOI: 10.1038/nrg.2017.97.
- Doench JG, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262-7. DOI: 10.1038/nbt.3026.
- Sanson KR, et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun. 2018;9(1):5416. DOI: 10.1038/s41467-018-07901-8.
- Lennox KA, et al. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016;44(2):863-77. DOI: 10.1093/nar/gkv1206.
- Khan AA, et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol. 2009;27(6):549-55. DOI: 10.1038/nbt.1543.
- Rayner E, et al. CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. CRISPR J. 2019;2(6):406-16. DOI: 10.1089/crispr.2019.0006.
- Schwertman P, et al. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat Rev Mol Cell Biol. 2016;17(6):379-94. DOI: 10.1038/nrm.2016.58.
- Zhu S, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol. 2016;34(12):1279-86. DOI: 10.1038/nbt.3715.
- Chavez A, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12(4):326-8. DOI: 10.1038/nmeth.3312.
- Stopka T, et al. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. 2005;24(21):3712-23. DOI: 10.1038/sj.emboj.7600834.
- Jost M, et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol Cell. 2017;68(1):210-23 e6. DOI: 10.1016/j.molcel.2017.09.012.
- Datlinger P, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14(3):297-301. DOI: 10.1038/nmeth.4177.
- Savell KE, et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro. 2019;6(1). DOI: 10.1523/ENEURO.0495-18.2019.
 3")

: A How-To Guide 5")



