

Signaling pathways are critical for proper cellular function, from controlling cell proliferation to triggering necessary cell death. Errors in signaling play a significant role in many diseases, including cancer and genetic disorders, and can be useful drug targets. However, identification of these potential drug targets is challenging, as signaling pathways are complex and involve a plethora of proteins and molecules.
CRISPR Screening
In the last few years, CRISPR (Clustered, Regularly Interspaced, Short Palindromic Repeats) screening methods have become successful additions to the drug discovery toolkit. [1] The CRISPR–associated protein 9 (Cas9) is a well-established system that manipulates individual genes in a targeted manner using guide RNAs (gRNAs).
Scaling CRISPR methods through the creation of gRNA libraries enables large genome-wide screens that provide an overview of all the genes involved in a specific pathway. [2] Both CRISPR loss-of-function (LOF) and gain-of-function (GOF) screens are possible at the whole genome level. [3] Ultimately, this can allow for unbiased identification of novel drug targets. [1] LOF screens can be achieved in two ways:
- Traditional CRISPR knockout (KO), where the target gene is directly edited to prevent the expression of a functional gene product.
- CRISPR interference (CRISPRi), where transcription of the target gene is inhibited by preventing the transcriptional machinery from expressing the gene. This method utilizes a catalytically inactive Cas9 and no gene-editing occurs.
CRISPR KO screens are more effective than RNA interference (RNAi) screens in ensuring complete gene knockdown. Additionally, CRISPR KO screens may lead to fewer off-target effects than RNAi screens. [4] RNAi controls gene expression at the mRNA level, while CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) regulate gene expression primarily on the transcriptional level. This makes it possible to target non-coding regions using CRISPRi and CRISPRa.
General Considerations for CRISPR Screening
There are five main aspects to consider when planning a CRISPR screen, summarized in Figure 1. Taking these aspects under consideration before screening can help in the identification of the most suitable libraries, format, and overall approach.

1. Reagents and Supplies
The first consideration when planning a CRISPR screen concerns the materials needed to carry out the screen. The availability, cost, and scalability of reagents and cellular models may impact the design of a screen. For example, the large numbers of cells required for screening may not be feasible if using primary cells, in which case a cell line with high levels of proliferation may be preferable. Considering the cost and availability of reagents is essential, as this may impact the scope of the screen. If reagents are abundant, a whole-genome screen may be plausible, whereas if reagents are a limiting factor, a smaller, more targeted screen may be more cost-effective.
2. Stable Cell Line Production
The first step of many CRISPR screening workflows involves the generation and isolation of cells expressing the Cas9 nuclease. This step can vary in the time and processes required to generate depending on the chosen cell line and whether transient or stable transfection is used. Stable expression of Cas9 ensures nuclease levels are consistent across all cells within a screen. However, generating Cas9 lines is more time consuming than using transient expression. For both transient and stable transfection, the isolation of cells successfully expressing Cas9 can be performed by either antibiotic selection or fluorescence-activated cell sorting (FACS), depending on whether an antibiotic resistance gene or fluorophore is present in the expression vector.
3. Drug Selection
The mechanism of action for the drug selected for the screen determines the required screening time as well as dictates the phenotype to screen. The phenotype, as discussed below, will influence the choice of screening platform.
4. Enrichment/Phenotyping
Consideration should be given to the measured phenotype, and clear expectations for the outcome should be set based on the results of small-scale preliminary experiments. Pooled screens will require the detection of a phenotype that allows for enrichment, such as cell proliferation or activation of a reporter-based assay. [2]
5. NGS and Deconvolution (For Pooled Screens)
When planning a pooled screen, an additional sequencing and deconvolution step is required to identify the positive hits, which requires access to a sequencing facility and bioinformatics support. Alternatively, Sigma-Aldrich® deconvolution services are a cost-effective way to identify essential genes from a pooled CRISPR screen.
Overview of Primary Workflows
After isolation of cells expressing Cas9, the cells are then transfected with the gRNA library before stimulation with the compound of interest. After the screen is complete, the data is analyzed, and any potential hits are validated.
The specific steps of a CRISPR screen differ depending on whether a pooled or an arrayed library is used (Figure 2).
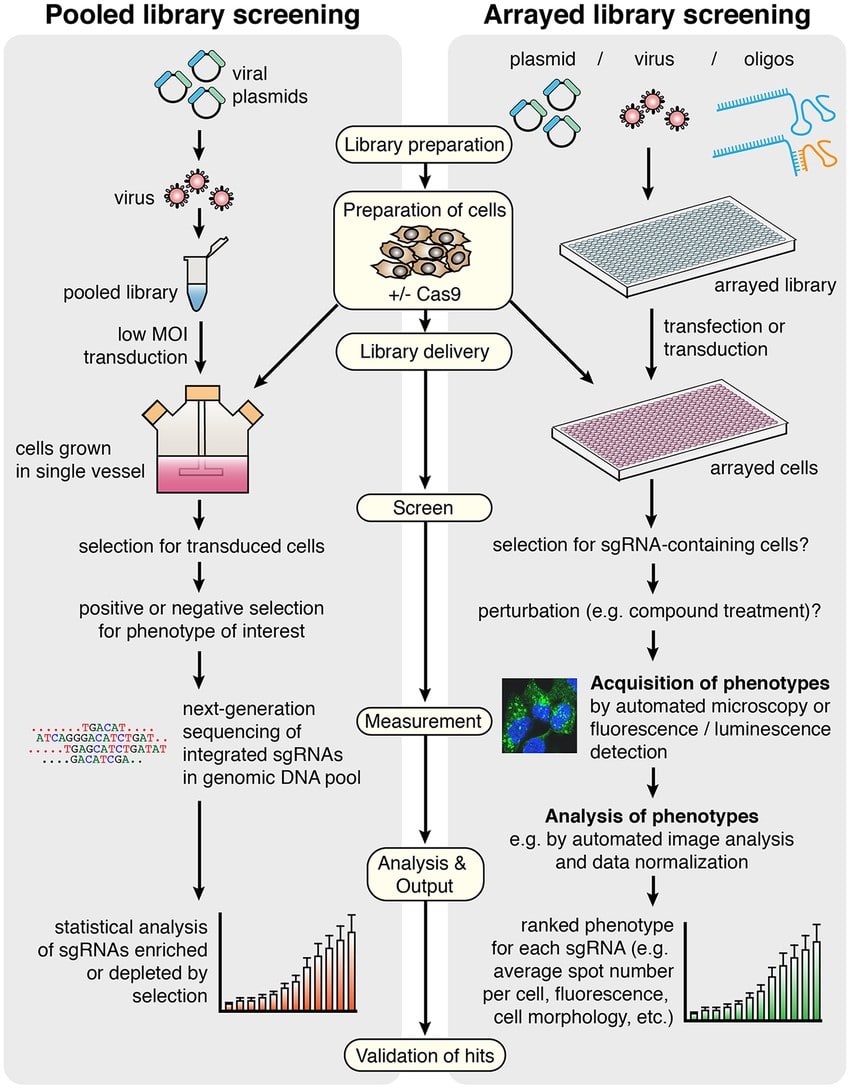
Pooled Library Screens
A pooled library can include thousands of gRNAs in the same tube, which are all delivered simultaneously to cells grown in a single vessel. This step requires permanent integration (e.g., through lentiviral transduction) into the cell genome to ensure the persistence of gRNAs that are later amplified to identify positive hits during deconvolution. [2]
Pooled libraries such as GeCKO or Sigma whole-genome pools provide a cost-effective and straightforward approach for whole-genome CRISPR screens, both in vitro and in vivo. [6] However, as the screen occurs in one single population of cells, it is more complicated to link phenotypes to gene alterations. Positive hits are collected by gene set enrichment, which limits the phenotypes assessed (e.g., proliferation or expression of a reporter gene). [7, 8] After enrichment, a deconvolution step is required to identify additional positive hits, which makes the analysis step more complicated compared to arrayed screens.
Arrayed Library Screens
In arrayed libraries, cells are grown in multi-well plates. Only one gene is targeted per well through the transfection of either a single or multiple gRNAs. After the screen, the phenotypic assessment of each well individually provides a simple way to identify hits. [9] This method also allows for a wide variety of complex phenotype readouts through high-content imaging or cellular phenotyping. [5] However, the arrayed approach requires access to automation equipment that can handle the large number of plates needed to cover all the targets of interest. With this method, screening time is also directly dependent on the number of clones, making arrayed library screens more feasible for interrogating a subset of genes than for the analysis of the whole genome.
Post-screen Considerations
Some considerations to keep in mind for the post-screening process include not only the mechanism of hit identification but also the subsequent validation steps. When using pooled screens, positive hits should be reanalyzed using additional gRNAs against the identified targets and further validated by analyzing protein expression and carrying out detailed characterization of the phenotypes. [7]
CRISPR Screening Formats
CRISPR screening can be performed in a variety of formats, many of which are amenable to scaling for screening applications. Common CRISPR formats include CRISPR knockouts (KOs), CRISPR knock-ins, CRISPR activation (CRISPRa), and CRISPR interference (CRISPRi).
CRISPR KO Screens
In a CRISPR KO screen, Cas9 introduces double-strand breaks at locations specified by gRNA. When the endogenous non-homologous end-joining (NHEJ) DNA repair system corrects these breaks, this often leads to the introduction of frame-shift mutations that effectively knock out the gene. [2]
Sigma-Aldrich® products include lentiviral vector-based CRISPR whole-genome KO libraries either as arrayed formats, such as the Sanger whole-genome library, [9] or as a pooled library such as the GeCKO and Sigma pools.
CRISPR Knock-in Screens
CRISPR knock-in experiments, which involve the insertion of a sequence into a target gene, can be used to introduce single nucleotide polymorphisms, transgenes, tags, or fluorescent reporters. However, achieving a CRISPR knock-in is more challenging than a KO as it involves providing a donor DNA template in addition to the CRISPR machinery. Therefore, this method is currently not easily scalable beyond single gene targets.
CRISPRa and CRISPRi Screens
Both CRISPRi or CRISPRa are scalable and allow screening of genes through both repression and activation of transcription. [10] These systems use a nuclease-deficient version of Cas9 (dCas9), which can still bind and recruit additional domains to a gRNA target site but cannot cut the target DNA. [3, 11] In CRISPRi, dCas9 is fused with a transcriptional repressor, such as KRAB, while CRISPRa requires transcriptional activators, including VP64. [11]
The most effective CRISPRa system uses the CRISPR Synergistic Activation Mediator (SAM) complex. This system uses a dCas9-VP64 complex, with two additional transcriptional activators introduced into the cell alongside the gRNA. [11, 12, 13]
Synergistic Use of Screens
Utilizing multiple screening formats provides complementary insights into the same pathways. [3, 10] One published example shows the benefit of CRISPR in researching cancer. In this example, a tumor growth screen using CRISPRa identified tumor suppressors of K562 cells, while a LOF CRISPRi screen in the same system detected essential housekeeping genes. [3] Likewise, CRISPRa offered additional information about toxicity pathways when complemented by a CRISPRi toxicity screen. The combined use of CRISPRa and CRISPRi identified both genes that sensitize and desensitize K562 cells to ricin treatment. [3]
Library Selection
Choosing the right library for a CRISPR drug screen can be challenging. Sigma-Aldrich® CRISPR products make it easier to select a suitable library by offering a range of choices. Through a collaboration with the Sanger Institute, Sigma-Aldrich® CRISPR screening products include a whole-genome arrayed library for CRISPR KO and arrayed library panels that can be delivered in plasmid or lentiviral format. For pooled screens, GeCKO or Sigma whole-genome pools, a human kinase pool, and custom CRISPR panels are available. Single-cell analysis is also possible with 10x Genomics compatible CRISPR pools.
CRISPR Drug Screening Summarized
CRISPR screening uses the CRISPR-Cas9 system with a gRNA library to modulate the expression of many genes at a time. CRISPR allows both LOF (CRISPR KO or CRISPRi) and GOF screens (CRISPRa). Both CRISPRi and CRISPRa use a dCas9 fused to either transcriptional repressors or activators.
The combined use of LOF and GOF screens makes it possible to identify both repressing and activating components of the studied pathways, allowing for the identification of genes that either mediate the downstream effect of a drug or feature resistance to certain drugs. This combined synergistic approach facilitates the identification of potential new targets for further investigation.
CRISPR screens are available as arrayed or pooled screens. Arrayed screens, while resource-intensive upfront, are simpler to analyze and can monitor complex phenotypes. Pooled screens are more cost-effective and simpler to carry out experimentally, but can assess only a small range of phenotypes and the downstream data analysis can be complex as deconvolution is required.
Sigma-Aldrich® CRISPR screening products include a variety of pooled and arrayed CRISPR libraries covering both whole genomes or gene panels and offer both of LOF or GOF screens. Additional services and support are available to researchers who would like to learn more about which screening tools are most useful for their research needs.
More CRISPR Resources
CRISPR Articles
- CRISPR Gene-editing: Considerations and Getting Started
- How to Understand CRISPR Formats and Their Applications
- How to Validate a CRISPR Experiment
- A CRISPR Approach to Immuno-Oncology
CRISPR Webinars
- A guide to leveraging single cell CRISPR screens for deeper biological insights
- CRISPR Screening: from design and discovery to validation
- Engineering Vero Cell Lines Using CRISPR to Increase Production of Viral Vaccines
- Efficient Generation of Gene-edited Mouse Models and Cell Lines Using Synthetic sgRNA
CRISPR eBook
CRISPR Tools
And you should also consider the following Sigma-Aldrich® resources:
Discover more about CRISPR in the Bitesize Bio CRISPR Research Hub.
References
- Fellmann, C., et al. Cornerstones of CRISPR–Cas in drug discovery and therapy. Nat Rev Drug Discov. (2017) 16, 89–100. DOI: 10.1038/nrd.2016.238
- Doench, J. G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Gen. (2018) 19, 67–80. DOI: 10.1038/nrg.2017.97
- Gilbert, L. A., et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. (2014) 159, 647–661. DOI: 10.1016/j.cell.2014.09.029
- Smith, I., et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLOS Bio. 15, e2003213 (2017). DOI: 10.1371/journal.pbio.2003213
- Agrotis, A., et al. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Front Genet. (2015) 6. DOI: 10.3389/fgene.2015.00300
- Chen, S., et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. (2015) 160, 1246–1260. DOI: 10.1016/j.cell.2015.02.038
- Miles, L. A., et al. Design, execution, and analysis of pooled in vitro CRISPR/Cas9 screens. FEBS J. (2016) 283, 3170–3180. DOI: 10.1111/febs.13770
- Yang, J., et al. Genome-Scale CRISPRa Screen Identifies Novel Factors for Cellular Reprogramming. Stem Cell Reports. (2019) 12, 757–771. DOI: 10.1016/j.stemcr.2019.02.010#
- Metzakopian, E., et al. Enhancing the genome editing toolbox: genome wide CRISPR arrayed libraries. Sci Rep. (2017) 7, 1–9. DOI: 10.1038/s41598-017-01766-5
- Horlbeck, M. A., et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife. (2016) 5, e19760. DOI: 10.7554/eLife.19760
- Konermann, S., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. (2015) 517, 583–588. DOI: 10.1038/nature14136
- Chavez, A., et al. Comparison of Cas9 activators in multiple species. Nat Methods. (2016) 13, 563–567. DOI: 10.1038/nmeth.3871
- Tragante V, et al. Gene Set Enrichment Analyses: lessons learned from the heart failure phenotype. BioData Min. (2017) 10:18. DOI: 10.1186/s13040-017-0137-5




