

The primary components of the CRISPR gene editing system are a bacterially-derived nuclease (e.g. Cas9) and a guide RNA (gRNA). This relatively simple system has proven to be incredibly versatile. Through a number of modifications, researchers have developed modified CRISPR nuclease formats that can be used for a variety of studies including gene knockout/knock-in, activation, repression, and epigenetic modification. The result is a toolbox that enables scientists to perform a breadth of gene editing and modulation experiments beyond the reach of systems like ZFNs or TALENS.
Here we review the general CRISPR/Cas system, discuss two of the most important alternative formats – nickases and catalytically dead nucleases – and their applications in research.
The CRISPR Nuclease System
The CRISPR system comprises a gRNA and a Cas nuclease, which together form a ribonucleoprotein (RNP) complex (Figure 1). This RNP targets and cuts specific DNA sequences. The specificity of the RNP is provided by the gRNA, which consists of two parts:
- CRISPR RNA (crRNA), which contains the protospacer; a 17-20 nucleotide sequence that is designed to be complementary to the desired target DNA and directs RNP-DNA binding specificity.
- Trans-activating crRNA (tracrRNA), which binds to the Cas nuclease.
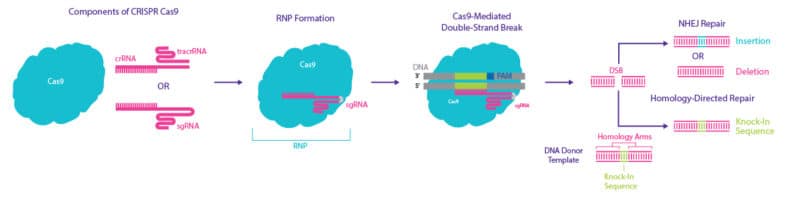
Although the gRNA is required to direct the RNP complex to the target DNA, it is not sufficient for successful binding and cleavage of the DNA by the RNP complex. The presence of a short sequence of DNA, known as a protospacer adjacent motif (PAM), is also required downstream of the target sequence. In the presence of this PAM, the RNP complex binds and, through the action of the nuclease, cleaves the target DNA creating a double-strand break (DSB).
Various CRISPR nucleases have been identified, with each recognizing a different PAM. The most commonly used, and most widely available, nuclease is the “wild type” Cas9 derived from the bacterium Streptococcus pyogenes (SpCas9). SpCas9 recognizes a PAM of 5’-NGG-3’ directly downstream of the target sequence, on the non-target strand of the genomic DNA
In cases where a suitable PAM for SpCas9 is not available for the desired target sequence, a Cas9 derived from a different species may be necessary. Several different Cas9 versions have been isolated from other bacterial species and these have been found to recognize different PAMs. For example, the Cas9 isolated from Streptococcus canis (ScCas9) recognizes a less stringent PAM of 5’-NNG-3’. [1] Cas9 isolated from Francisella novicida (FnCas9), like SpCas9, recognizes a PAM of 5’-NGG-3’ but with a preference for A, T, or C in the ‘N’ position. In contrast to SpCas9, the DNA cleavage performed by FnCas9 is staggered, which results in 5’ overhangs after the DBS.
Modified CRISPR Nuclease Formats
CRISPR Nickases
One limitation of wild type Cas9 variants is that they have a low stringency for DNA complementarity, which can result in off-target effects. To compensate for this, Cas9 nickases have been engineered that create single-stranded breaks (nicks, which can be repaired by the cell’s machinery) rather than double-stranded breaks. For example, researchers have introduced a D10A point mutation that inactivates the RuvC nuclease domain in SpCas9, so that the engineered nickase can only cleave the target strand [2].
Therefore, in order to generate a double-stranded break (DSB), two different, but adjacent, gRNAs are used with paired nickases (Figure 2). This approach effectively removes the possibility of off-target effects, since both Cas9 nickases must nick their targets to generate a DSB.

Moreover, when two Cas9 nickases are used, instead of blunt-ends, cohesive ends are produced providing even greater control over gene insertion and integration. This feature makes CRISPR nickases an ideal system for performing gene editing in therapeutic applications. In fact, nickases are already being used for genome editing in primary T cells as well as excision of viral DNA (e.g., hepatitis B virus, HBV) in humans [3].
Nuclease-deficient Cas9
The nuclease-deficient version of Cas9 (dCas9) has been engineered to be catalytically inactive. While dCas9 cannot cleave DNA, it maintains the ability to precisely target DNA, allowing it to serve as a cargo-delivery system to specific sequences in the genome. Current uses for dCas9 range from gene activation and interference to epigenetic modification. Here we explore some of these implementations
CRISPR Activation
CRISPR activation (CRISPRa) uses a modified CRISPR-dCas9 system, which includes transcriptional activators attached to either the dCas9 or the gRNA to increase the expression of a target gene. Most CRISPRa systems use dCas9 fused to the polymeric viral transactivation domain, VP64 (Figure 3). The synthetic transcription factor is then targeted to a specific promoter sequence by the gRNA. Coupling the RNP with additional coactivators, such as p65 and HSF1, further increases the potency of the CRISPRa system [4].
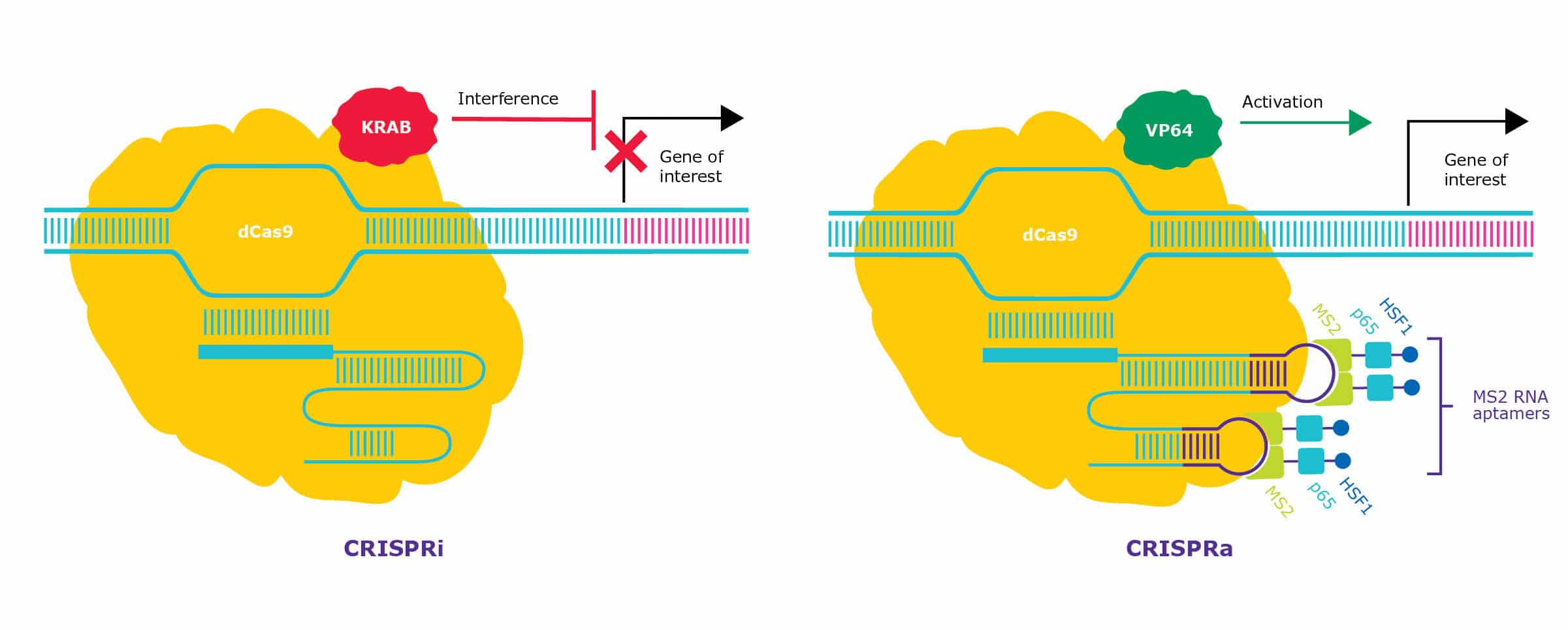
One example of CRISPR gene activation is called CRISPR SAM (synergistic activation mediator). This platform combines VP64-dCas9 with a modified guide. The gRNA contains two aptamers that recruit additional co-activators, p65 and HSF1, enhancing target gene activation (see Figure 3). This SAM has been shown to increase the expression of difficult targets like OCT4 hundreds of folds [5].
CRISPRa can also serve as a complementary approach to loss-of-function (LOF) genetic screens by enriching for a different set of genes responsible for a phenotype [3]. CRISPRa screens show little-to-no off-target activity and can be used to study genes that are difficult to characterize using only LOF approaches because of their high copy number. This topic is covered in more detail here.
CRISPR Interference
CRISPR interference (CRISPRi) provides a highly efficient method for silencing genes without altering the underlying DNA sequence. The CRISPRi system is based on a dCas9 fused to a transcriptional repressor, such as the well-characterized Krüppel associated box (KRAB) domain (see Figure 3). KRAB recruits proteins that result in epigenetic repression surrounding the target DNA (e.g. DNA methylation). When targeted to promoters of genes, this activity prevents the recruitment of the transcriptional machinery, effectively silencing gene function.
The KRAB-dCAS9 CRISPRi system has been used to successfully silence genetic components in non-dividing neurons, which are notoriously difficult to edit at the genomic level [6]. CRISPRi exhibits better targeting specificity compared to other conventionally used gene silencing approaches such as RNA interference (RNAi) [7].
CRISPR Epigenetic Modifiers
In addition to expression modulation, CRISPR-Cas9 can also be used to make epigenetic modifications surrounding the targeted DNA, allowing researchers to study the epigenome directly. Epigenetic effectors, such as histone acetyltransferases, can be targeted to genes of interest in order to study the epigenetic control of these loci as well as induce or repress expression in a more native manner.
One example is the dCas9-p300 CRISPR gene activator system. This uses the catalytic histone acetyltransferase domain of the human p300 protein to unwind DNA, thereby allowing for endogenous activation of gene expression. In practice, OCT4 transcript levels have been increased at least two-fold when activated using p300-dCas9 [8].
CRISPR-based epigenome modification can also be utilized in high-throughput screens to identify and study functional regulatory elements for specific loci [9].
Summary of Modified CRISPR Nuclease Formats
The CRISPR gene-editing system provides a range of tools to study gene expression in several complementary ways. Engineered versions of Cas9 have allowed the potential off-target effects of wild type Cas9 to be effectively reduced or eliminated (via eSpCas9 or Cas9-nickases) and have specific utility for nuclease-independent regulation of gene function.
In addition, dCas9, when coupled with epigenetic modifiers, has opened up the epigenome to direct interrogation and manipulation.
If you want to know more about the CRISPR system, you can find additional information in our related CRISPR resources.
More CRISPR Resources
CRISPR Articles
- CRISPR Gene-editing: Considerations and Getting Started
- How to Validate a CRISPR Experiment
- Level Up Your Drug Screening With CRISPR
CRISPR Webinars
- Engineering Vero Cell Lines Using CRISPR to Increase Production of Viral Vaccines
- Efficient Generation of Gene-edited Mouse Models and Cell Lines Using Synthetic sgRNA
- CRISPR Screening: from design and discovery to validation
CRISPR eBook
CRISPR Tools
And you should also consider the following Sigma-Aldrich® resources:
- Advanced Genomics Resource Center
- CRISPR Lentiviral Formats
- CRISPR Controls
- CRISPR activation
- Cas9 Protein Comparison
- Sigma-Aldrich® Advanced Genomics
- Additional CRISPR Webinars
Discover more about CRISPR in the Bitesize Bio CRISPR Research Hub.
References
- Chatterjee, P. et al. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci Adv. Oct; 4(10) (2018). DOI: 10.1126/sciadv.aau0766
- Lamberth, J. et al. Highly efficient and specific genome editing in human cells with paired CRISPR-Cas9 nickase ribonucleoproteins. BioRxiv (2019) DOI: 10.1101/766493
- Karimova, M. et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci Rep 5, 13734 (2015). DOI: 10.1038/srep13734
- Chavez, A. et al. Comparison of Cas9 Activators in Multiple Species. Nat Methods 13 (7), 563-567 (2016). DOI: 10.1038/nmeth.3871
- Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583-8 (2015). DOI: 10.1038/nature14136
- Zheng, Y. et al. CRISPR interference-based specific and efficient gene inactivation in the brain. Nat Neurosci 21, 447-54 (2018). DOI: 10.1038/s41593-018-0077-5
- Kampmann, M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem Biol 13 (2), 406-416 (2018). DOI: 10.1021/acschembio.7b00657
- Ji, Q. et al. Engineered zing-finger transcription factors activate OCT4 (POU5F1), SOX2, KLF4, c-MYC (MYC) and miR302/367. Nucleic Acids Research 42(10), 6158-67 (2014). DOI: 10.1093/nar/gku243
- Klann, T. et al. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol 35, 561–568 (2017). DOI: 10.1038/nbt.3853





