
Experimentally determining the structure of a protein is very difficult. Why bother with it then?
It may sound flippant, but you don’t always have to.
There are so many experimental protein structures now that the non-experimental structures of a protein can usually be modeled using experimental structures of similar proteins as templates. The most common method used to do this is called homology modeling.
Don’t get me wrong, an experimental structure is always superior to a model, but:
Choose a free resource to help you move forward
CHEAT SHEET
Western Blot Cheat Sheet
DOWNLOAD
Western Blot Troubleshooting Card
- A protein may belong to a class that’s notoriously difficult to solve;
- You need specific expertise and equipment to solve the structure of a protein;
- Even with that expertise and equipment, a structure solution project may fail.
So, in this article, we’ll cover homology modeling, the principles behind it, and some useful tools that can help you perform it. We’ll also take a brief look at some alternative modeling methods and where they fit in.
Meet the Candidates
There are 3 main methods of modeling a protein structure:
- Homology modeling; [1,2]
- Threading/fold recognition; [3]
- Ab initio modeling. [4]
Methods 1 and 2 provide three-dimensional structure predictions of proteins using experimental structures as templates. The templates will have been obtained via X-ray crystallography, NMR Spectroscopy, and cryo-electron microscopy. These methods also take into account a number of physical energy functions.
Method 3 doesn’t require a template structure. Hence the name. Ab initio means “from the beginning.”
Of the 3 methods, homology modeling is the star attraction.
This is due, in part, to the increasing availability of a large number of experimentally determined protein structures. Most genomes on the tree of life have a representative number of published experimental structures.
That being said, experimental structures of membrane proteins are far fewer than water-soluble proteins. (Of the 183,980 structures in the Protein Data Bank, 5354 are membrane proteins at the time of writing.) The issue of membrane protein structure solution remains a “frontier” in biology and is a field in which modeling is highly beneficial.
Here are a few more details on the three modeling methods.
1. Homology Modeling
You only use this method when there is a protein with an experimental structure available that you suspect resembles your target protein (say over 30% sequence identity).
It relies on programs such as BLAST [5] to search for similar proteins in various databases, structural or otherwise, such as the Protein Data Bank (PDB). Your homology model is then constructed from the suite of search results.
Homology modeling is also called “comparative modeling,” because you’re comparing the model structure with known template structures as you build it.
2. Threading/Fold Recognition
With this method, you predict the structure of your target protein using known protein folds for similar proteins found in various different databases. You can easily generate models using the online server I-Tasser. [6]
3. Ab initio Methods
This method predicts protein structures when experimental structural information of similar proteins is not available. The model is built from scratch by calculating the most favorable energy conformations of the amino acids.
This method should only be used as a last resort since results from it are less reliable than template-based methods.
The 8 Steps of Homology Modeling
At the outset, it should be noted that there are web-based servers for automated homology modeling. Protein Homology/analogY Recognition Engine V 2.0 (PHYRE2) [7] and SWISS-MODEL [8] are excellent examples.
Upload a primary protein sequence, set a few parameters, and off you go. Your structure prediction will be with you in a few hours.
These homology modeling servers are very sophisticated and generally produce excellent results. Check out figure 1 for a good example of the kind of result you can expect.
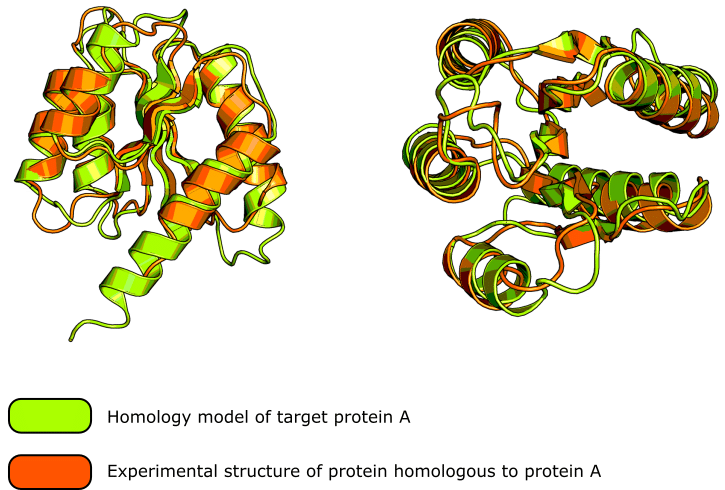
If, however, you’re going to perform homology modeling manually, then consider the process as a ladder with rungs on it. Each rung, or step, is important in its own way and we cannot skip a step and jump up.
If we do then, our final structure may turn out to be grossly incorrect and lead to erroneous interpretations.
You’ll need some software for manual homology modeling. A good example is MODELLER. [9] Note that this is an open-source Python script. A GUI version of the program, called Easy Modeller, is available and is simpler to use. [10]
Oh, and if you like the protein cartoons in figure 1, be sure to read this article on creating figures in PyMOL™ to learn how to generate them.
1. Target Sequence Selection (Optional)
This step depends upon your needs. The protein sequence you wish to model is termed the “target sequence.” Do pick the appropriate length of target protein that allows you to just focus on the bit you’re interested in.
That’s to say, in some cases, you don’t need to model an entire protein. And in such cases, sticking with the essential protein sequence/domains will save you work and speed things up.
2. Template Protein Recognition
The template proteins are the reference protein structures. In homology modeling, you match your target protein with all of the related protein structures that are present in the various databases using simple sequence alignment software. Select the proteins that are most closely related to your target to use as template structures.
3. Preparation of Template Protein (Optional)
Now, you have to trim back the template proteins because the experimental structures will contain extraneous matter. For example, symmetry equivalent protein chains, water molecules, ligands, and solvent of crystallization.
4. Sequence Alignment
Next, you need to align your target and template protein sequences using a sequence alignment tool such as CustalW. [11] This is a very important step in homology modeling because using an appropriate alignment algorithm is necessary for bagging the most valid template structures.
The alignment compares all of the proteins, target, and template(s), and can tell you which parts have completely conserved amino acid sequences.
5. Prediction of Secondary Structure
The secondary structure of the model is built using tools present in ExPASy Portal. It compares the proposed secondary structures of your target to those of the template proteins and ranks them to iteratively build up the model.
6. Tertiary Structure and the Tentative Model
With the secondary structure elements of the model built, you can start to piece these together and predict the tertiary structure of your target. You then need to visualize the model and check its overall structure to make sure it’s sensible.
7. Loop Modeling
If any loops are present in the model, you can optimize them further if they have unrealistic conformations. Use loop modeling software such as Modloop [12] to do this and improve the quality of your final model.
8. Model Optimization and Validation
Finally, once you have an almost complete model, you’ll need to improve it to attain a near-native confirmation via energy minimization. You can do this using protein model validation tools and verification servers. Such validation tests show whether your protein model is energetically satisfactory based on the spread of conformations observed in experimental structures for any given fold or feature. [13]
Be Aware of the Limitations of Your Model
We’re all standing on the shoulders of giants, and homology modeling needs a lot of shoulders. Remember that a homology model is only indirectly based on experimental data. The model itself has no experimental evidence to back it up.
Remember also that homology modeling can only exist because of the work of crystallographers, NMR spectroscopists, and microscopists. So if you’re collaborating with one and their experimental structure ends up disagreeing with the homology model. Tough.
Reality will never be as neat and tidy as anything that’s generated in a computer.
Also, be mindful of how well-represented the protein you’re modeling is. If you suspect it adopts a fold that has been deposited time and time again in the PDB, that’s great. Your model is probably going to be quite reliable.
If, however, you’re modeling a protein that you suspect belongs to a class of (structurally) under-represented proteins, be duly cautious with the implications that you draw from your homology model.
Observations should always be weighed against the number and quality of data.
Hmm… What to Do with Your Protein Model?
The outputs from homology modeling are used in a variety of applications across a broad range of sciences, including:
- Molecular Interaction Studies such as Molecular Docking;
- Computer-aided drug discovery;
- Improving Quantitative Structure-Activity Relationship (QSAR) results;
- Overcoming the phase problem in protein crystallography. [14]
A Note on DeepMind
Any article discussing modeling would be remiss not to mention AlphaFold at DeepMind. This sophisticated AI tool is providing exquisite predictions of protein structures from one-dimensional amino acid sequences. Go and check it out if you haven’t already, and note that the structures can be accessed for free!
Has this article helped you to understand homology modeling? Would you like to see any other aspects of homology modeling discussed on Bitesize Bio? Comment below if so!
Originally published December 2017. Reviewed and republished on November 2021.
References
- Krieger E, Nabuurs SB, and Vriend G (2003) Homology modeling. Methods Biochem Anal 44:509–24
- Rodriguez R et al. (1998) Homology modeling, model, and software evaluation: three related resources. Bioinformatics 14:523–8
- Rost B, Schneider R, and Sander C Protein fold recognition by prediction-based threading. J Mol Biol Jul 270:471–80
- Yuan X, Shao Y, and Bystroff C (2003) Ab initio protein structure prediction using pathway models. Comp Funct Genomics 4:397–401
- Altschul SF et al. (1990) Basic local alignment search tool. J Mol Biol 215:403–10
- Roy A, Kucukural A, and Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–38
- Kelley L, Mezulis S, and Yates C et al. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–58
- Waterhouse A et al. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
- Šali A and Blundell T (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Bio 234:779–815
- Kuntal BK, Aparoy P, and Reddanna P (2010) EasyModeller: A graphical interface to MODELLER. BMC Res Notes 3:226
- Thompson JD, Higgins DG, and Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–80
- Fiser A and Šali A (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19:2500–1
- Hooft RW, Sander C, and Vriend G (1997) Objectively judging the quality of a protein structure from a Ramachandran plot. Bioinformatics 13:425–30
- Evans P and McCoy A (2008) An introduction to molecular replacement. Acta Crystallogr D Biol Crystallogr 64:1–10
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.



 4")

 Screening 6")
