
These days it seems AI is everywhere! Whether writing articles or analyzing complex data sets, AI is changing everything—it’s even changing the somewhat niche field of protein structure science.
Specifically, AI has become extremely powerful at accurately predicting protein structure via either ab initio structure modeling or protein structure prediction. Even better, these AI tools are free and open access!
Historically, getting a protein structure for your research was an arduous process, which could be a limiting step for your research; but not anymore!
What tools are out there, and how do they work? What are their respective strengths and weaknesses? What does protein structure prediction mean for experimental methods like NMR and X-ray crystallography?
Choose a free resource to help you move forward
EBOOK
Free Guide to Protein Expression
DOWNLOAD
Western Blot Troubleshooting Card
In this article, we’ll answer these questions.
Overview of Protein Structure Prediction
Proteins, the workhorses of the cell and one-time contenders for being the molecule of genetics until DNA came along, often tantalize researchers, with the structural science falling short of being able to predict a protein’s structure based solely on the amino acid sequence.
If you have a stretch of genomic DNA sequence, you can predict:
- where the introns are,
- where transcription will start and stop,
- where translation will start and stop,
- and even predict distal regulatory elements and methylation sites.
Try to predict protein tertiary structure, or what residues affect enzyme activity with just a string of amino acids, and historically, you would have come up short. This is known as the protein folding problem.
But, with new protein structure prediction tools, that may be changing! Check out Figure 1 to see a predicted protein structure and how well it compares to the experimental structure of a putative structural homolog.
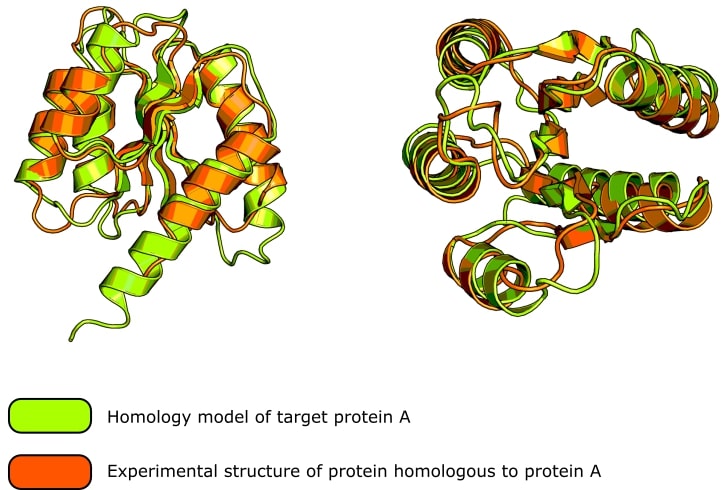
Figure 1. The green protein is a homology model of the N-terminal domain of AgrA, a response regulator found in Staphylococcus aureus. The orange protein is the crystal structure of LytR (also from S. aureus, PDB: 6m8o), which has a 34% sequence identity to protein AgrA and is a suspected structural homolog. The homology model, generated using PHYRE2, predicts protein AgrA to exhibit the same fold as LytR. The overall root mean square deviation between the two structures is 1.07Å. (Image credit: Thomas Warwick.)
A Very Brief History of Protein Structure Prediction Tools
There have been many milestones in structure prediction, starting from the first Ramachandran plot and the first homology-based structure prediction from an amino acid sequence in the 1960s. Developments then brought us software like MODELLER, and SWISS-MODEL in the late 1990s, and now the AI protein structure prediction methods like AlphaFold-2.
Hundreds of thousands of structures have been experimentally determined. This means that the protein’s structure was determined using laboratory techniques like X-ray crystallography, NMR, or cryo-electron microscopy to derive the 3D position of every atom in the protein.
And after a structure is determined, it can be visualized in, and downloaded from, the Protein Data Bank (PDB). Structural science has progressed a long way, and if you are interested in a comprehensive timeline of protein structure prediction methods, check out this review by Pearce and Zhang, who do a great job of summarizing the last 50 years of achievements. [1]
Now let’s explore whether AI protein structure prediction methods really have solved the protein folding problem.
Why Should I Use AI Tools to Predict Protein Structures?
As with most things in biology, to truly understand function, you need to understand structure. So, there’s one answer—to understand function.
A Protein’s structure is also useful when discovering drugs. For instance, if you wanted to inhibit an enzyme involved in some disease state, knowing the shape of that enzyme’s active site or an allosteric region (a distal region of the structure that impacts the shape of the active site from afar) could go a long way toward the design of a specific inhibitor.
Or, maybe your interests are more basic. Perhaps you are interested in how alternatively spliced transcripts influence the corresponding proteins’ structure, or maybe you want to determine how two or more proteins fit together inside the cell — questions that complement experimental approaches like chromatin conformation capture techniques for probing spatial organization.
The benefits of knowing a protein’s structure are almost too numerous to count, but protein purification and growing crystals can be extremely difficult, labor-intensive, and in some cases, damn near impossible — and even when purification succeeds, quality control of your purified protein matters before any downstream work.
Running an amino acid sequence through software is a much better option, and what I mean by this is that you are predicting the arrangement of a protein’s atoms in 3D based on predictive algorithms and inference alone. No experimental data is used to generate the structure of the protein.
Sounds great!
But can you trust the software to get it right?
To properly consider if these tools are accurate and trustworthy, we’ll need to understand how these protein structure prediction tools work.
How do Protein Structure Prediction Tools Work?
Homology-Based Structure Prediction
One way to predict the structure of your protein is to compare its amino acid sequence to another protein with a solved structure—a process known as homology-based structure prediction. If the sequences are similar, it stands to reason that their structures should also be similar.
If the amino acid sequence homology between the template protein and your protein is very high, you can simply superimpose the side and main chain atoms onto the known structure and derive the structure of your protein (see Figure 1).
If there are some differences in amino acid sequence, you can superimpose the main chain atoms onto those regions and manually determine where the side chains will go. Once you have a preliminary model based on sequence homology, you can refine it to ensure that the confirmation—things like the bond angles and energy minimization of folds—makes theoretical sense.
Threading
Another approach is called threading. [2] Here, you do not overlay an amino acid sequence to a homologous structure, but instead, you take existing structures and see if your sequence could potentially match their folding.
There are only so many possibilities for protein conformations in nature, and even proteins that lack sequence homology to one another may have similar three-dimensional structures.
So, you pick several candidate templates for threading and use an algorithm to determine which template results in the best fit, looking at compatible bond angles and the lowest energy score. This process is iterative and is a good option if a protein structure with a homologous sequence does not exist.
AlphaFold 2
The next approach, made possible by modern computing power and AI, made a huge splash during the 14th Critical Assessment of Structure Prediction (CASP14) assessment in 2020. Specifically, I’m talking about AlphaFold-2, which was co-developed by DeepMind. [3]
This method starts by running a multi-sequence alignment (MSA) that considers the evolutionary relationships between proteins and, thus, changes in individual amino acids. For instance, if a given residue has mutated, then another amino acid “paired” to that residue will also change so that the protein’s overall structure is maintained in the variant.
The alignment and pairings are iteratively passed through a machine learning algorithm AlphFold-2 refers to as an evoformer. This algorithm identifies the best pair interactions and alignments and passes the information to a third portion of the pipeline that generates a structure. The last two parts are repeated three times, generating the final predicted structure.
The AlphaFold-2 development team ran the sequences of proteins with experimentally solved structures through the AI pipeline and found that the predicted protein structures were highly similar to the experimentally determined ones. [3]
Briefly, from the CASP14 challenge, AlphaFold-2 was able to predict the coordinates of backbone atoms in space with an accuracy of 0.96 Å root-mean-square deviation (RMSD), and an all-atom accuracy of 1.5 Å RMSD. To put this in perspective, the width of a carbon atom is 1.5 Å, and the all-atom accuracy of the next best approach entered in CAPS14 was 3.5 Å RMSD. [3]
The deviation of atomic coordinates by less than 1.5 Å would result in actual and predicted structures that are very nearly superimposed upon each other!
I know what you are thinking right now—this sounds too good to be true, and we can say goodbye to the art of growing protein crystals. There must be a catch.
What are the Valid Uses of Predicted Protein Structures?
What you choose to use AlphaFold or any other in silico protein modeling tool for, and whether it’s a sensible choice or not, really depends on what you want to do with the prediction itself.
For instance, if you wanted to narrow down potential binding partners to a given protein based on their structures, then prediction tools could be very useful, especially if you plan to validate these functional predictions experimentally. See Table 1 for some ideas to get you thinking about how predicted structures can aid your experimental work.
Table 1: Ideas for how structure prediction can aid your research.
| Application | How it helps your research |
| Protein mutagenesis | Introducing targeted mutations to ablate binding to some other protein without completely destroying all the functionality of the mutant would take a lot of trial and error if you knew nothing about the protein’s structure. In silico predictions could narrow down candidate mutations by predicting the resulting structure post-mutagenesis. |
| Function of alternative splicing variants | Almost all genes encoding proteins produced alternatively spliced versions. How do these structures compare to the full-length version for which you already may have structural and functional information? Predictions may help you understand how the splicing variants function in vivo. |
| Screening ligands | This one speaks for itself if you have hundreds or thousands of candidate ligands to screen against your protein. This can narrow down candidates to a manageable number for testing experimentally. |
What are the Drawbacks and Pitfalls of Predicted Protein Structures?
While sequence homology modeling and threading have their uses, they rely on comparisons. Additionally, although AlphaFold-2 can predict structures with incredible accuracy, it is not 100% certain.
So, if you want the protein’s structure solely for its own sake, and it’s super important that the structure is accurate, then you will need to consider if prediction tools are really a valid and sensible substitute for traditional techniques such as crystallography, NMR, or cryo-electron microscopy.
In other words, if your conclusions are based solely on a protein’s structure, and you cannot support these conclusions with any sort of experimental work, you will naturally want to use a model with very high accuracy to avoid erroneous conclusions.
That means you will have to do some research on how protein structure prediction methods work, if there are proteins structurally similar to yours, and if so, how many. A protein structure with dozens of published structural homologs, then you probably don’t need to solve it yourself experimentally.
Note also that AlphaFold-2 does have some difficulty predicting the structure of antibodies [4], and it has some trouble with intrinsically disordered proteins. [5] It also cannot model allostery, which is usually essential in drug discovery. [5]
So, if any of these examples were important to your work, you would definitely want to support conclusions based on structural predictions with additional experimental work, even including determining the protein’s structure the old-fashioned way!
Structure Prediction Tools for Your Research
If you are convinced that computer and AI-based protein structure predictions are for you, here are some tools and resources you can use in your work.
Tools for predicting protein structures:
- Check out this excellent YouTube video explaining how AlphaFold-2.
- Oxford Protein Informatics Group has a blog dedicated to protein informatics, including several posts on AI and AlphaFold-2.
- AlphaFold Protein Structure Database (hosted by EMBL-EBI), where you can get access to millions of structure predictions made by AlphaFold.
- Protein Structure Prediction Center. Host of the CASPs and contains a wealth of information on structure prediction methods.
- Code for AlphaFold made available by DeepMind.
- PHYRE homology modeling tool.
- Automated modeling server.
Summary
Protein structure prediction is a dynamic and active field. Work continues on AI-powered methods for predicting protein structure, and DeepMind has made the code for AlphFold-2 publicly available to allow others to make their own modifications to the pipeline.
Currently, the results from AlphFold-2 are impressive, and researchers around the world are experimenting with it. Indeed, AlphaFold, combined with other AI tools in biology, has a lot of potential to advance research, but time will tell whether we can say goodbye to growing protein crystals in the lab!
References
- Robin Pearce R and Zhang Y. (2021) Toward the solution of the protein structure prediction problem. J Biol Chem 297(1)
- Bowie JU, Lüthy R, and Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253(5016):164–70
- Jumper J, Evans R, Pritzel A, et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–89
- AlphaFold 2 is here: what’s behind the structure prediction miracle. Oxford Protein Informatics Group. Accessed 19 Feb 2023
- Nussinov R, Zhang M, Liu Y, and Jang H. (2022) AlphaFold, Artificial Intelligence (AI), and Allostery. J Phys Chem B. 126(34):6372–83
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.







