
Adapted from Precise, High-Efficiency Editing of Stem Cells to Probe Human Biology and Model Disease, a BitesizeBio Webinar with Bill Skarnes.
Studying human diseases and genetic variation isn’t easy. In vitro studies allow us to manipulate the genome of cells reasonably easily, but creating model systems that are easy to work with and give valid data is challenging.
To overcome this, we usually resort to immortalized cells. But these cell lines are not normal. They have significant mutations on their genome, exhibit attributes not found in healthy in vivo cells, and some have unethical origins.
Fortunately, there’s an alternative to immortalized cells.
Choose a free resource to help you move forward
EBOOK
Gene Editing 101
DOWNLOAD
Blood Collection Tube Chart
Using CRISPR to edit induced pluripotent stem cells is a way to create physiologically relevant and translational models. That’s what we’ll discuss in this article.
We’ll make the basics extremely clear and explain the guiding principles of genome editing topics. Plus, we will give you a heap of practical steps to monitor and improve editing efficiency, design guide RNAs, control the zygosity of your clones, and more!
The CRISPR iPSC Genome Editing Platform to Probe Human Biology
This practical approach to exploring human biology and modeling disease combines two technologies that have won Nobel Prizes. These are:
- Cell reprogramming.
- CRISPR.
We’ll delve a little deeper into these next. But briefly, cell reprogramming enables the creation of induced pluripotent stem cells (iPSCs) from somatic cells, and, using CRISPR, we can edit the genome of iPSCs to change their phenotype.
This gives us access to a vast amount of human biology. For example:
- Early human development (embryoids).
- Organogenesis (organoids).
- Hematopoiesis/innate immunity.
- Rare diseases.
- Gene therapy.
- Cell biology.
- Metabolomics.
Now let’s examine these technologies in a little more detail.
Induced Pluripotent Stem Cells
The power of iPSCs is that they can be differentiated into almost any cell type of the human body.
Plus, these cells have normal genomes, unlike cells commonly used in the past to address human cell biology and biochemistry, such as HeLa and HEK293 cells, which have highly abnormal genomes that do not represent that of the average person.
And genomic abnormality aside, the non-consensual origin of HeLa cells raises a serious ethical issue.
Furthermore, you can culture iPSCs indefinitely and maintain a normal diploid karyotype: a healthy set of 23 homologous pairs of chromosomes.
There are some disadvantages to iPSCs. The main ones are that the efficiency of inducing pluripotency is sometimes low and involves the viral delivery of sometimes carcinogenic genes into somatic cells.
For a closer look at the advantages and drawbacks of iPSCs and how they are prepared, check out this paper by Medvedev, ShevchenkI, and Zakian. [1]
What Is CRISPR?
CRISPR is a revolutionary gene editing technology that needs little introduction to most readers. If you need an introduction, check out Bitesize Bio’s easy guide to CRISPR.
In short, it’s a powerful tool to cut and alter DNA sequences both in vitro and in vivo. A small RNA molecule called guide RNA (gRNA) guides a nuclease (usually Cas9) to a desired target location in the genome to remove or add genes, correct mutations, and even regulate gene expression.
CRISPR offers two salient advantages over conventional gene editing tools. These are:
- You can make single base pair edits by cutting or nicking DNA. [2]
- You can modify both copies of a gene simultaneously (biallelic mutations).
However, for CRISPR to be a valid genome editing tool, there are two essential requirements:
- The target sequence intended for modification is genomically unique.
- The target sequence is several base pairs upstream of a protospacer adjacent motif sequence (a PAM sequence).
If either of these criteria is not met, CRISPR will not work.
Check out this post to learn more about Cas9s and what strands they nick.
What Are PAM Sequences?
PAM sequences are short sequences of DNA that are essential for cleavage by a Cas nuclease.
They are located on the non-complementary strand: the nucleic acid strand, with the same sequence as the guide RNA.
And they are typically 3–4 base pairs downstream of the Cas nuclease cut site.
You’ll find a list of common PAM sequences in Bitesize Bio’s guide to nucleases and towards the bottom of this Addgene article on CRISPR.
A word to the wise—when designing guides for your CRISPR experiments, avoid ones that introduce mutations into the PAM sequence.
Practical Guidance for CRISPR iPSCs Genome Editing Projects
Now you know the benefits of creating an alternative to immortalized cells using iPSCs and CRISPR, here are some practical tips on how to do it well.
Guiding Principles
Five basic principles or steps that apply to any genome editing project.
1. Subclone the iPSCs
Subcloning induced pluripotent stem cells (iPSCs) enables you to isolate a clonal population of cells with the best genetic properties for your project.
Doing so will reduce the genetic heterogeneity of your edited clones, increasing the consistency and reproducibility of your results.
Furthermore, subcloning helps eliminate undesirable off-target effects that may arise from the genome editing process.
2. Careful Characterization of the iPSCs
It’s always wise to characterize your iPSC line to ensure they have:
- An intact genome. Use karyotyping, high-density SNP arrays (discussed later), and whole-genome sequencing.
- Unmutated TP53. It often becomes mutated in cultured iPSCs.
- Good differentiation potential. Use an mRNA microarray, RNA sequencing, or qPCR. [3]
3. Edit the Genome
Introduce mutations that relate only to your study: ones found in the human population with no additional changes.
Although you can introduce silent mutations which improve editing efficiency, these could impact gene transcription and cause unwanted off-target effects.
4. Characterize the Post-Edit Clones
Characterize your post-edit clones to check if you have introduced your desired mutation.
Also, check the cells to ensure they retain a normal genome after editing. Look for off-target effects you may have accidentally introduced due to your desired mutation.
Karyotype the cells again and use long-range PCR to inspect high-density SNP arrays to determine the zygosity of your clones (more on this later).
5. Control/Reversion
Once you identify the cell phenotype you would like to prove is due to the mutation, not something else acquired during culturing and editing, revert it to the wild-type sequence using CRISPR (Figure 1).
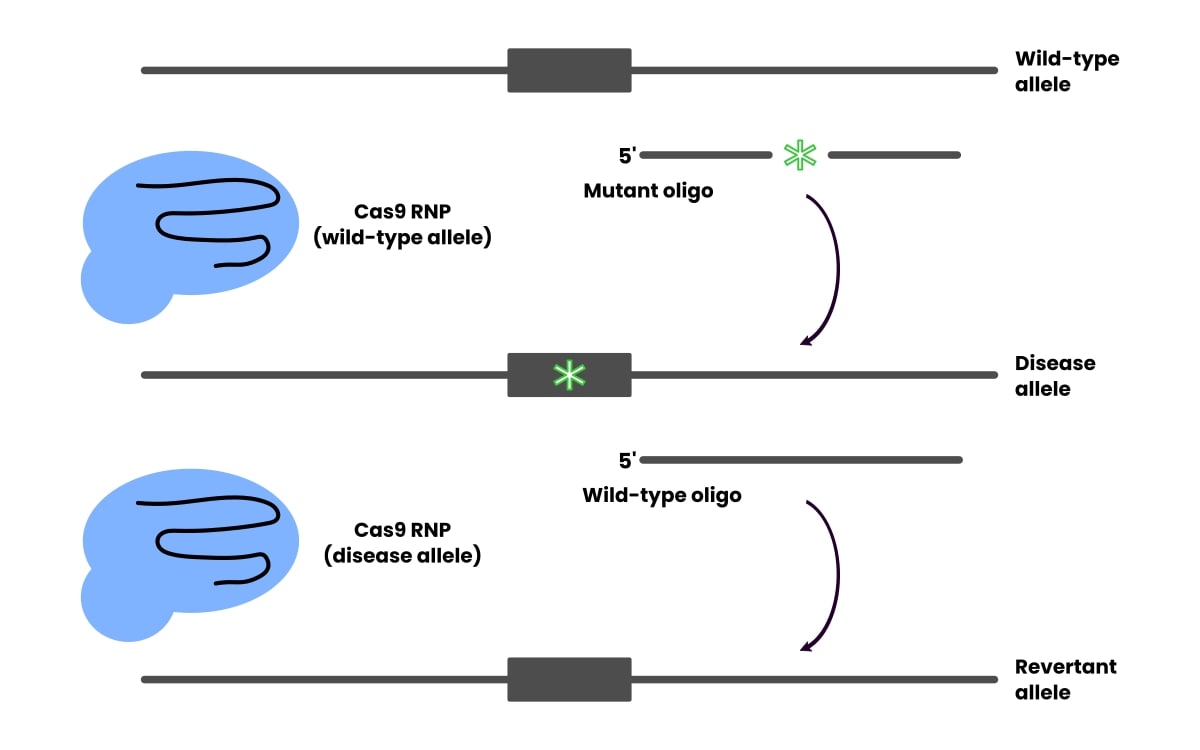
Reversion controls for any off-target effects, but be aware that the revision efficiency doesn’t necessarily correlate to the editing efficiency.
DNA Repair Methods in CRISPR
RNA serves as a guide to direct the Cas nuclease to specific target DNA sequences, and the RNA–nuclease complex is called a ribonucleoprotein (RNP).
These complexes recognize a specific side of the genome and make a double-stranded break.
You can use either of two pathways to repair the break:
- Non-homologous end joining.
- Homology-directed repair.
Non-homologous end joining (NHEJ) is an error-prone process that results in insertion/deletion mutations at the break site on one or both alleles. This is useful for projects where you want to mutate the gene in such a way.
Homology-directed repair (HDR) involves adding a donor template that will get incorporated at the breakpoint and introduce single nucleotide variants.
Check out Figure 2 to see this represented graphically.
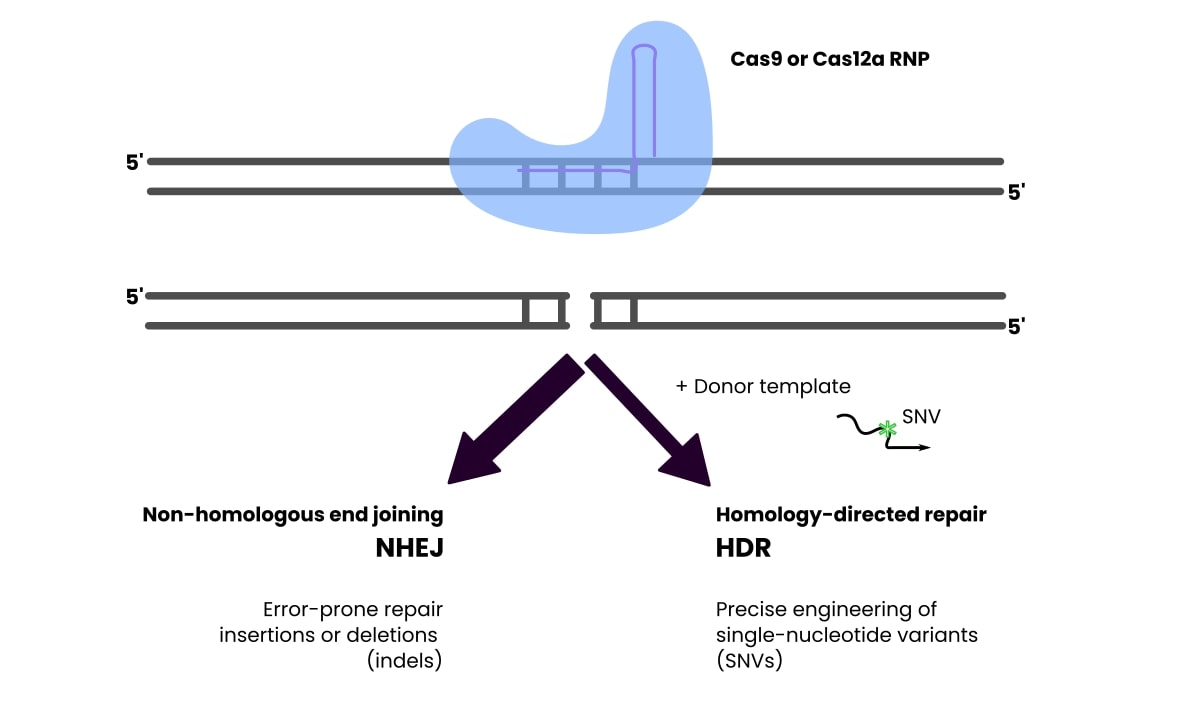
The two methods compete with each other. HDR is precise but can be inefficient, with a low percentage of the cells exhibiting the desired mutation. This could be a problem for large-scale projects.
NHEJ is more efficient but prone to the imprecise mutation types mentioned above.
Strategies to Improve the Efficiency of HDR
As mentioned, the nuclear infection efficiency for HDR can be low. Perhaps lower than 10%. Fortunately, there are ways to improve its efficiency. Strategies to increase its efficiency include:
- Small molecule enhancers. [4]
- Cold shock. [5]
- End-modified oligo donors. [6]
The benefit of these methods is they involve delivering reagents and conditions to the cells. No cellular selection or enrichment is needed.
And note that this list isn’t exhaustive. You can check out an overview of strategies here. [7]
How to Monitor the Efficiency-Improving Strategies
So you’ve got some ideas on improving your gene editing efficiency, but how do you monitor them to know if they have worked?
If you change the C to a T in the histidine codon of blue fluorescent protein BFP, you get green fluorescent protein GFP.
- CAT → Histidine.
- TAT → Tyrosine.
These proteins have different spectral properties; the former is blue, the latter green.
You can introduce this mutation using CRISPR under different iPSCs conditions to monitor the efficiency of the editing process and select conditions that maximize editing efficiency.
This is a simple yet powerful assay to monitor the efficiency of NHEJ and HDR. Plus, the results are easy to interpret! [8,9]
- No mutation → blue fluorescence.
- Successful HDR mutation → green fluorescence.
- Successful NHEJ mutation → no fluorescence.
The reason that NHEJ will result in no fluorescence is because of the insertion or deletion mutations. It can even result in frameshifts when the length of the insertion or deletion is not a multiple of three.
Designing Guide RNAs
When doing genome editing, you must design guide RNAs that get the Cas nuclease to the desired target site on the genome.
WGE is an excellent CRISPR guide RNA design tool that helps identify guides that overlap the target site and shows you their position relative to the PAM sequence. [10]
It also gives an off-target efficiency score that quantifies the number of off-target sites with X base differences.
Aim for guides that have no matches to other areas on the genome and very few one- or two-base mismatches.
And, as mentioned, avoid guides that mutate the PAM sequence.
Screen Guide RNAs in vitro and in vivo
Guides are the wild cards in genome editing experiments, and there will be variable activity between guides.
A guide may have an excellent off-target score but only return wild-type clones or work for only (say) NHEJ. An almost identical guide (say just offset a little bit to the first one) may return ample HDR base-edited cells with the desired mutation.
In most cases, several or more potential guide RNAs will guide the Cas9 nuclease to the target sequence for cleavage.
When this is the case, screen them in vitro using a mobility shift assay to see which guides cleave the target sequence best.
Then, when you come to use guides for genome editing experiments, tabulate your sequencing results to see which guides give you homozygous biallelic mutations, heterozygous mutations, and no mutations (wild-type).
You may also wish to test if the guides work for HDE, NHEJ, or both.
You may discover a guide will only work for one repair method. This will give you an insight into how well a given mutation is tolerated or if it is lethal. For example, no mutations were introduced via HDR, but you get abundant wild-type and successful NHEJ-edited clones.
How to Control the Zygosity of Clones
Depending on what you are studying (especially if you are working on genetic diseases), you may wish to control the zygosity of your edited clones.
In other words, control whether you introduce the mutation to one or both alleles.
An effective strategy might be to add inactivated Cas9 (also called dead Cas9 or dCas9) with the guide RNA during the nuclear infection step. [9]
The dCas9 will bind to the target sequence, but there will be no cleavage.
So the dCas9 protects the cleavage and modification site from active Cas9 on any given gene copy.
Note that the amount of dead Cas9 you add is critical to the outcome. So add a mixture of Cas9 and dCas9 to your experiments. For example, 1:1, 1:1.5, 1:2, 1:4, ratios, etc.
Then, measure the dose response of your cells to dCas9, and plot the percentage proportion of homozygous, heterozygous, and wild-type clones against the dose to pick the sweet spot for your desired outcome.
You can also monitor and quantify this optimization using the BFP assay mentioned above because the dCas9 will increase the proportion of wild-type clones, leading to less conversion of BFP to GFP.
Quality Control: Check for Homozygous Clones
Deleterious on-target effects that can occur during the editing process must be screened for.
For example, one allele contains the desired mutation, while the other is lost altogether.
To check whether a homozygous clone is truly homozygous, you can do long-range PCR, inspect heterozygous single nucleotide polymorphisms (SNPs) on either side of the target sequence, and check if they are still heterozygous after you have made your edit. [11]
Loss of SNP heterozygosity indicates that one allele has been deleted or translocated. Or there is an unwanted insertion.
So beware!
The CRISPR iPSC Alternative to Immortalized Cells In Summary
That’s a lot to summarize, but I’ll try!
Significant problems arise when using immortalized cells, but CRISPR and cell reprogramming combine to create an alternative to immortalized cells.
We’ve explored the power of this technology and how to use it to study human disease and genetic variation.
And readers involved in existing genome editing projects now have an arsenal of ideas, tips, assays, and sources, to add extra layers of control.
Here’s to more impactful results and discoveries from them.
Have you got anything to add? Let me know in the comments section below.
Want more information? Watch the full presentation below.
References
- Medvedev SP, Shevchenko AI, and Zakian SM (2010) Induced pluripotent stem cells: problems and advantages when applying them in regenerative medicine. Acta Nat 2(2):18–28
- Rees HA and Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19(12):770–88
- Liu LP and Zheng YW (2019) Predicting differentiation potential of human pluripotent stem cells: Possibilities and challenges. World J Stem Cells 11(7):375–382
- Yu C, Liu Y, Ma T, et al. (2015) Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16(2):142–7
- Guo Q, Mintier G, Ma-Edmonds, M, et al. (2018) ‘Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci Rep 8:2080
- Ghanta KS, Chen Z, Aamir Mir, et al. (2021) 5′-Modifications improve potency and efficacy of DNA donors for precision genome editing. eLife 10:e72216
- Liu M, Rehman S, Tang X, Gu K, Fan Q, Chen D, and Ma W (2019) Methodologies for Improving HDR Efficiency. Front Genet 9:691
- Skarnes WC, Pellegrino E, and McDonough JA (2019) Improving homology-directed repair efficiency in human stem cells. Methods 164–165:18–28
- Skarnes WC, Ning G, Giansiracusa S, et al. Controlling homology-directed repair outcomes in human stem cells with dCas9. bioRxiv
- Hodgkins A, Farne A, Perera S, Grego T, Parry-Smith DJ, Skarnes WC, and Iyer V (2015) WGE: a CRISPR database for genome engineering. Bioinformatics 31(18):3078–80
- Weisheit I, Kroeger JA, Malik R, Klimmt J, et al. (2020) Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep 31(8)
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.






