
What do DNA mini preps and protein immunoprecipitation experiments have in common? They start differently, but they end with the same, critical stage – elution. But what exactly is elution, and what is the point?
The Terminology
First, let’s start with some basic terminology:
Elution – extracting one material from another by washing with a solvent.
The adsorbent – a solid phase, which can be a silica gel in the case of mini-prep columns, but usually beads that can be covalently linked to antibodies or other ligand molecules. “Solid phase” does not necessarily mean a standing column; it can be beads in an eppendorf that are easy to wash.
Choose a free resource to help you move forward
download
The Molecular Cloning Cheat Sheet
CHEAT SHEET
Nuclear Extraction Protocol
Affinity – a measure of the ability of the absorbent to bind the molecule of choice (what you are trying to elute). The higher the affinity of the solid phase to the biomolecule of choice (BOC), the tighter the molecule binds to it. However, you don’t want the binding to be irreversible; this will make the elution impossible.
Eluent – a solvent that removes BOC from the absorbent.
Eluate – the solvent containing BOC removed from the adsorbent.
Material Preparation
Before the elution, you need to absorb the molecule of choice while getting rid of the contamination. This is an essential step, as the conventional wisdom reminds us “garbage in, garbage out.” You may have excellent reagents for elution but if your sample contains too much of the unrelated staff (the scientific term is “gunk”), it will clog the adsorption material. Solid phase saturation will prevent your BOC from absorbing and then contaminate the eluate. The effective lysis and clean-up steps are essential for the success of your elution experiment.
It is important to determine the volume of your pre-absorption material. The volume of lysate that passes through the absorption medium should not exceed 3 – 5 volumes of the column. The large volume of lysate that passes through the absorbent increases the time of the experiment, as well as the likelihood of gunk absorption. In many cases, it’s worthwhile to reduce the initial volume of lysate by filtration or fractionation. Thus, the lysate volume determines the size of the column.
The choice of adsorption material depends on the chemical composition of your molecule of interest. The absorption of biomolecules usually involves more or less specific interaction between the substrate and the molecule. For example, DNA is absorbed onto mini-columns due to ionic interaction between negatively charged DNA phosphate groups and positively charged silica particles.
Proteins are usually adsorbed on sepharose or magnetic beads covered in IgG.
After an initial lysate application, at no point your column can dry out. This will “bake your” molecule to the absorbent and disrupt the column integrity. If you don’t have time to continue the experiment, top up the column with a compatible buffer and stop the flow.
Washing
The purpose of washing the solid phase is to remove a non-related material, while leaving the molecule of interest on the column. The selective separation is often achieved by using a buffer with low ionic strength (e.g. low salt concentration). The volume of the washing buffer should be close to the amount of the initial material and be at least 3-5 volumes of the column.
However, after several volumes of the washing buffer passed through the column, the contaminations will be washed away and any additional washing will not improve the quality of your prep. Moreover, you will start losing your target material.
Elution
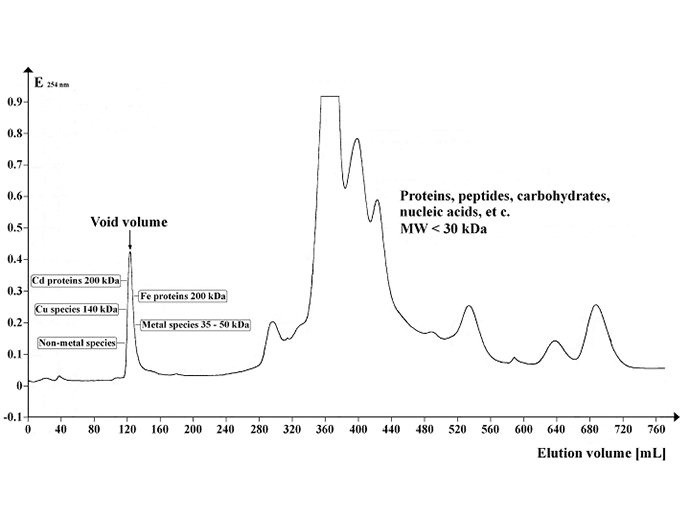
Elution itself works because you disrupt the bonds between the column and the substrate (i.e. by using high salt or high temperature of the eluent). Elution is usually done in a small volume of buffer compatible with sample storage and further applications.
DNA elution from the mini-prep column is the simplest case: one volume of buffer removes almost all DNA. The concentration of DNA in the eluate is inversely proportional to the elution buffer used: the more buffer you use, the less the final DNA concentration. However, even in this case, most manufacturers recommend using an additional volume to remove all DNA.
For the columns, elution speed is critical. Too slow speed will increase the chances of molecule degradation; too fast and there will be no fractions resolution.
For large volume columns, you need to collect eluate fractions because your molecule will be distributed between them. The first fraction will contain a mix of washing and elution buffer and possible contamination not removed by the washing buffer.
You can monitor OD for your type of molecule (260nm/280nm for DNA) and do a blot for your specific molecule concentration in each fraction. In the simplest case, your molecule distribution will follow a simple bell curve, but it can have one or more sharp peaks.
In conclusion, knowing the basic parameters of your experiment (absorbent, size of the column, washing buffer, elution buffer, flow rate, number of the fractions) and general principles of elution will allow you you to set up your elution successfully.
For more details, find an article where the other scientists did something similar – ideally the same molecule, but a similar will do – and adapt to your condition.
Sources:
- Elkins KM (2013). “DNA Extraction“. Forensic DNA Biology. pp. 39–52. doi:1016/B978-0-12-394585-3.00004-3. ISBN 9780123945853.
- Scopes RK (1994). Protein Purification – Springer. doi:1007/978-1-4757-2333-5. ISBN 978-1-4419-2833-7.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.






