
Studying nucleic acid interactions with proteins can be accomplished using a rapid and efficient electrophoretic mobility shift assay (EMSA). This method is essentially an agarose gel electrophoresis technique that detects protein:nucleic acid interactions, as the mobility of the labeled nucleic acid will be retarded if bound to a protein (compared to unbound DNA).
A lesser-known technique is the PEMSA, or protein electrophoretic mobility shift assay, which uses the polyacrylamide gel electrophoresis (PAGE) method and is more like a redox (reduction-oxidation reaction) western blot. If you want to be able to determine redox modifications to the cysteine (Cys) residues found in your protein of interest, or if certain conditions alter the protein redox state, then PEMSA might work for you! For example, you may want to know why your transcription factor is not binding to a DNA promoter, or why you cannot activate a certain pathway. Just like phosphorylation signaling cascades, which is a more common post-translational modification, redox modifications can cause activation/deactivation of pathways. There are even antibodies that can detect various phosphorylated proteins, but detecting redox modified proteins is a bit more involved as discussed below.
Getting Started
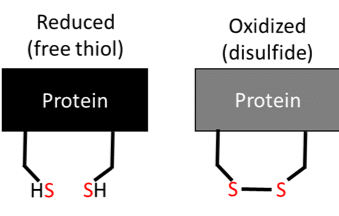
PEMSA is a useful method to determine whether a certain condition is affecting the redox state of your proteins and therefore if the Cys residues are reduced or oxidized (Figure 1). These thiols can be modified in a number of different ways, but this article will focus on whether the thiol is reduced (SH), or oxidized and exists in a disulfide bond (S-S) with another Cys (Figure 1). These different ‘species’ of Cys can be “labeled” with charges that will then separate out the reduced Cys from the oxidized Cys residues using the PAGE method.
Setting up the Samples
Choose a free resource to help you move forward
DOWNLOAD
Western Blot Troubleshooting Card
EBOOK
Free Guide to Protein Expression
Reagents
To denature the proteins for downstream “labeling”, you can either use urea or guanidinium HCl in the buffer. In my experience, guanidinium HCl is easier to work with and use it instead of urea.
For alkylating agents, I use iodoacetic acid (IAA) as well as iodoacetamide (IAM). The IAA adds a negative charge to the reduced Cys residues (-SA–), while IAM adds a neutral charge to the reduced Cys residues. As the samples run toward the positive electrode, the protein Cys labeled with IAA (and thus the negative charge) will migrate faster on the PAGE, compared to IAM labeling (neutral charge). Reduction and alkylation methods are commonly employed for identification of peptides for mass spectrometry, as well.
In order to label the Cys residues, they need to be in the reduced state. I use dithiothreitol (DTT) to reduce the samples. This may sound counter intuitive, since the goal is to be able to discern the reduced from oxidized Cys residues. However, that is why we use two different labels: the Cys that are naturally reduced at baseline are labeled with IAA (negative charge), and the Cys resides that are naturally oxidized are reduced and then labeled with IAM (neutral charge).
I also use diamide, which is an oxidant and is a useful control for the sample. After treating a sample with increasing concentrations of diamide, under denaturing conditions I alkylate the sample with IAA which labels reduced thiols. The reason for this is that thiols could be buried in the protein and protected from oxidation. I would then treat the sample with a reductant, and label with IAM to separate the Cys that were initially oxidized from the reduced Cys at baseline.

Optimization
To make sure protein charge labeling is correct, concentrations, incubation times, and temperatures may need to be optimized for each of the above-mentioned reagents. For example, you may want to add DTT and then split the sample, labeling one half with IAA and the other with IAM. Make sure the IAA sample runs farther than the IAM labeled samples.
I make a mobility standard, or a “ladder” to represent the reduced Cys in the protein, the oxidized Cys in the protein, and any disulfides or combination of species that might exist as shown in Figure 2. A protein with 2 Cys may contain mixed disulfide states in which 1 Cys may be reduced and the other Cys is bound to a substrate. These can be represented by the multiple bands shown in Figure 2.
To make the ladder, divide the protein lysate in denaturing buffer into 3 aliquots after reducing with DTT. Add IAA to one sample, IAM to another, and a ratio of IAA- IAM to the last aliquot and incubate for 30 minutes. Mix the aliquots together to generate one sample of all of the charged isoforms for the Cys in the protein of interested. Again, this might need to be optimized for proteins with differing numbers of Cys residues.
Remove unreacted IAA
Remove the unreacted IAA using a sephadex spin column, as it is a little less time consuming then precipitating the proteins with cold acetone, which can also be done. I think for MS, quenching the unreacted IAM is necessary, but I have not come across a protocol that does this for the PEMSA method.
Preparing the Gel
I use a homemade discontinuous polyacrylamide native gel consisting of a 5% stacking gel and a 15% running gel. I can usually make up several of these at once, but tend to try to use them within the same week.
What Could Go Wrong?
- Lack of optimization: It is important to optimize your reducing conditions to ensure all of the protein you are interested in is reduced and alkylated. If you have a lot of protein, you may need to add more reductant, or try a different reductant.
- IAA:IAM ratio is wrong: This is important so that you get all the different redox states for your protein.
- Collected samples too slowly: Redox reactions can be very fast, so it is important to optimize your collection time. I have collected samples as early as one minute!
- Gel wasn’t made correctly/transfer of gel: It is possible something went wrong with the actual making of the gel or transfer of the gel. APS, or ammonium persulfate is used along with TEMED to polymerize the gel, and that has to be made fairly fresh.
In order to make sure the reaction was complete, and I was able to alkylate and separate the reduced from oxidized Cys residues there was a bit of optimization I had to do. Once I prepared the diamide treated controls and optimized the mobility standards I did not have any trouble seeing reduced from oxidized Cys species. However, you may need to optimize the voltage and time at which the gel is run in order to separate and quantify the multiple bands. And lastly, make sure the samples did not run off the gel by using dye and that you have a good antibody to detect your sample. Most of the optimization involves the concentration, temperature, and reaction times for the alkylating and reducing agents.
References
- Bersani NA, Merwin JR, Lopez NI, Pearson GD, Merrill GF. Protein electrophoretic mobility shift assay to monitor redox state of thioredoxin in cells. Methods Enzymol. 2002;347:317-26.
- Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem.2003;278(35):33408-15.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.







