Western Blotting is a long established method for which the protocol varies little from lab to lab. However, there are kits available and some tweaks that can be made to the protocols that may improve your results and reduce the time it takes you to execute this popular technique.
Save Time by Co-Incubating the Primary and Secondary Antibodies
There are a number of kits that, I believe, are based on this simple concept. Normally, we incubate the blocked membrane with the primary antibody, wash away the unbound antibody, then incubate the membrane with the secondary antibody which binds to the primary antibody, co-localizing the detection method (fluorophores, horseradish peroxidase (HRP), or alkaline phosphatase (AP)) with the protein of interest on the membrane.
However, since the primary antibody binds its epitope with its variable domain and the secondary antibody with its constant domain, there is no reason that mixing the two antibodies together will interfere with the binding of the primary antibody to its epitope.
Both the primary and secondary antibodies are diluted into your favorite solution (TBST + 5% milk, for example) and incubated with the blocked membrane at the same time. For the average user, this will shave over an hour off a western blot by eliminating the second antibody incubation along with the washes between the two antibody incubations. I have used this method for around five years, and have yet to have any problems with it.
Put this article into practice
Choose a free resource to help you move forward

EBOOK
Free Guide to Protein Expression

DOWNLOAD
Western Blot Troubleshooting Card
Optimize the Incubation Times
If you find yourself doing the same western blot repeatedly (i.e. same primary and secondary antibodies with similar samples), then you might be able to shave even more time off the standard protocol.
A friend of mine experimented with the blocking time and incubation time with the primary/secondary antibody solution. She found that the blot was blocked after a 10 minute incubation with fresh, room temperature 5% milk, and that the western signal reached maximum after a 15 minute incubation with her antibodies (without increasing the concentration of the antibodies).
She could develop a blot and be ready for exposure within 45 minutes of removing the membrane from transfer. These parameters are going to vary for each set of antibodies and sample types that one uses, so you would need to decide if it is worth you time to systematically optimize these incubation times.
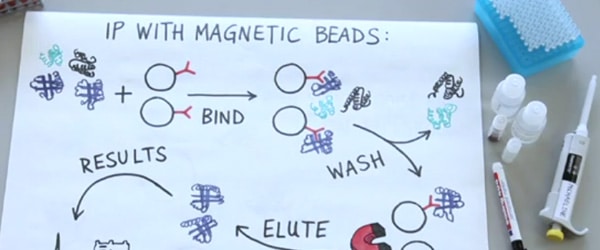
Use Protein A-HRP to Get Clean Western Blots of Immunoprecipitated Samples
If your immunoprecipitation (IP) used an antibody from the same organism as the primary antibody for your western blot, then your secondary antibody will not only recognize the primary bound to the epitope, but also the antibody from the IP that was denatured and separated out on the gel, creating giant background bands around 55 and 25 kD on the blot.
However, in 2005 Ashish Lal published a method to circumvent this problem: instead of using a secondary antibody to detect the location of the primary, use HRP conjugated Protein A instead. Protein G worked well, also, but did show higher affinity to the denatured antibody.
Unlike a secondary antibody, Protein A only recognizes correctly folded antibodies. This means that it binds tightly to the primary antibody used in the western, but largely ignores the denatured antibody stuck to the membrane. This system has (seemingly) been incorporated into several kits, but Protein A-HRP alone is also available from a number of companies.
Protein A-HRP can be used for normal western blots as well, and would replace secondary antibodies against Mouse, Rabbit, and Guinea Pig, which might be an attractive option for labs on a budget or with a fair number of undergraduates, who seem inexplicably drawn to the incorrect secondary antibody.
Multiplex Westerns
If you need to gather information about two different proteins in the same sample, you can probe the blocked membrane with an antibody against the first protein, then strip the blot and probe with an antibody against the second protein; run two identical gels and probe each with a different antibody; or if your two proteins are of very different sizes, cut one membrane in half across the lanes and probe the top and bottom sections with different antibodies.
Alternatively, you can perform a multiplex western.
A multiplex western is one in which more than one primary antibody is used on the same membrane at the same time. This obviously isn’t a complicated change to the protocol – simply add the extra primary antibody (and secondary, if needed) to the appropriate solution. However, I am mentioning it here simply because I have seen many occasions when people could have used this approach to save themselves time or sample, but didn’t.
If you are using chemiluminesence detection methods, then of course you need to have characterized the antibodies sufficiently to know that you are interpreting the western blots correctly. This can save you time in the day-to-day experiments, but for critical experiments (i.e. paper figures) I will still blot with the individual antibodies separately, just to make sure my interpretation is correct.
The multiplex western can be taken a step further if the two primary antibodies were raised in different animals. In the high-tech version of this technique, the two secondary antibodies are labeled with two different fluorophores, and, after washing, the bands are differentially detected using a Storm imaging system or equivalent.
In the low-tech version, one of the secondary antibodies is conjugated to HRP, while the other secondary is conjugated to AP. Each of these enzymes can be detected by chromogenic substrates, allowing you to develop a visible two color blot. The caveat to the latter system is that the chromogenic substrates are known to be less sensitive and reduced linearity of response compared to fluorescent or chemiluminescent methods.
The approaches that I’ve described here can help you streamline your westerns and save you some time, but I think it is important to stress that any changes to a protocol should only be implemented with the proper testing and controls. If you are happy with your standard protocol, then you shouldn’t change it. If you are like me and have found your work day extended into a work evening on a regular basis by the standard western protocol, then the suggestions above may help you keep your work hours under control without sacrificing your productivity.
Now to hear from you: what are your best western blot tips and tricks?
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.

Put this article into practice
Choose a free resource to help you move forward

CHEAT SHEET
SDS-PAGE Protocol Cheat Sheet
DOWNLOAD
Western Blot Troubleshooting Card