
As Morrissey once sang “in the midst of life we are in death, etc.” A fact of life in the research Lab is that whenever we run an assay there will almost certainly be some cell death in our sample. This may be insignificant in some techniques but in others it can be problematical.
Why dead cells are a problem
Flow cytometry is one technique where dead cells can be very problematic; this is because:
- they will take up the antibodies and probes that we use non-specifically
- they may become more autofluorescent.
Both of these consequences will cause an increase in background fluorescence and therefore reduce the dynamic range and reduce our ability to see something that is weakly positive for a marker. If that wasn’t bad enough, dead cells may release DNA which is notoriously sticky and will lead to cell clumping (not useful in a single cell technique like flow cytometry), and in cell sorting you really don’t want to compromise your experiment by sorting non-viable cells.
Solving the dead cell issue
However fear not, we can target these dead cells and remove them, if not from the tube, at least from the analysis. Firstly we can make sure that the medium we keep the cells in contains a small amount of DNAse which will help keep cells apart. Secondly we can use a feature of dead cells that will allow us to specifically identify them. When cells die, they lose their membrane integrity and become leaky. This is what causes us problems with non-specific binding but this time we are going to use a specific staining to help us identify those dead cells. There are two ways to do this – by using dyes that bind to DNA or dyes that bind to protein.
Choose a free resource to help you move forward
DOWNLOAD
Blood Collection Tube Chart
DOWNLOAD
The Happy Scientist Reference Pack
DNA binding dyes
Dyes such as Propidium Iodide (PI), DAPI, DRAQ7, 7-AAD and TO-PRO-3 will only enter cells whose membranes have been compromised. Once they do, they will bind to DNA and fluoresce so that these cells can easily be detected and gated out of any analysis (Figure 1A). In most cases the protocols are very simple, just add an appropriate amount of DNA dye to the sample about 10 minutes before running them on the cytometer (be aware of leaving the dyes on too long before analysis because every cell that dies when the dye is present will be positive so if you wish to compare between samples, keep the staining time consistent!). There are a range of these dyes with different excitation and emission characteristics so it shouldn’t be a problem to find one that will fit with your experimental panel.
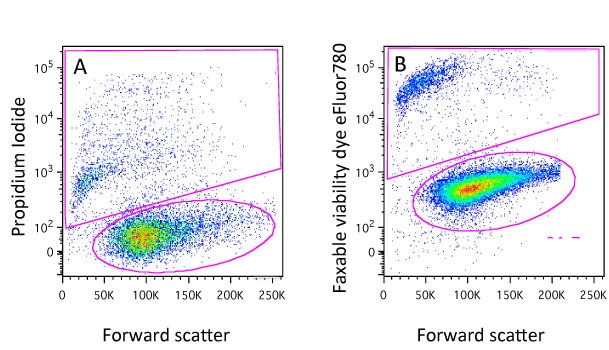
You will notice though that here we have added the dyes to live cells. If we were to add say PI to a samples of fixed cells, it would stain all the cells as fixation will permeabilise the cells. This is obviously a drawback if cells have to be fixed for safety reasons or because you want to detect an intracellular target. It will come as no surprise to learn that we have thought of that and this is where the protein-binding dyes come in…
Protein-binding dyes
These are also sometimes known as amine-reactive dyes or live/dead fixable dyes. They are still based on the principle that dead cells have compromised membranes but work in a slightly different way. Instead of binding to DNA they bind proteins so if they are added to a sample before fixation, live cells will bind some dye – there will be proteins present on the surface of the cell, but dead cells will bind much more as the dye can enter the cell and have access to the abundance of intracellular proteins.
At this point cells maybe fixed using with formaldehyde or alcohol and the difference in staining level will be maintained (Figure 1B). Again, protocols are very simple, a 20 minute incubation with the dye and these dyes come in a variety of excitation and emission characteristics and again you would almost certainly find one compatible with your other fluorochromes. Examples of theses dyes are the live/dead fixable dyes, the eFluor fixable dyes, the Horizon dyes, the Biolegend Zombie dyes and the Ghost dyes. Although the dyes can be fixed, this is not mandatory so they can also be used to identify dead cells in sorting experiments.
In these days of multi-laser flow cytometers there should never be any excuse not to add a viability dye and now you know how simple it is, you can do it too!
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.




