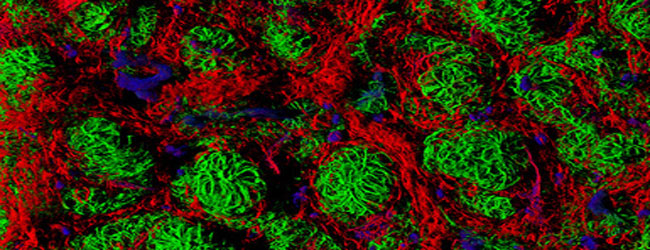
For a long time we’ve been able to pinpoint the subcellular location of proteins, and the advent of FISH (Fluorescence in situ Hybridization) allowed us to locate the position of genes in the nucleus, but recent advances in RNA FISH are making it easier and easier to collect the same data about individual messenger RNAs.
The Mechanism Behind FISH
For the uninitiated, fluorescence in situ hybridization is a method where specific DNA or RNA sequences are visualized inside a fixed and permeablized cell by annealing a labeled nucleic acid probe to the sequence of interest. Unlike proteins, where antibodies specific to the endogenous protein of interest have to be raised via random massive parallel screening in a biological system (in other words, squirt it in a bunny, mouse, goat, or undergraduate*, and screen the serum), we already have a method to generate a specific probe to a known sequence – synthesize the complementary sequence. This method was originally, and is most popularly, used against DNA targets. Given a sufficiently specific probe and appropriately stringent annealing and washing conditions, the FISH probe is much like a magnet used to find a needle in a real haystack: it is attracted to and binds only to its complementary sequence in the complex genomic haystack.
Standard RNA FISH
It wasn’t long before the methods that were developed for DNA FISH were applied to RNA and they worked (assuming you skipped the RNase step in the DNA FISH protocol). Some of the first RNA FISH experiments actually used labeled antisense RNA as a probe, however most labs switched over to using DNA probes developed for DNA FISH. There are several different approaches used to generate the labeled DNA probes used in FISH, but the two most popular methods are PCR and Nick Translation. In the PCR method, the probe is simply generated by PCR of the sequence of interest, doping the reaction with a dNTP covalently attached to a label, and incorporated into the DNA by the thermostabile polymerase. The Nick Translation method modifies a purified plasmid or DNA fragment containing the sequence of interest by replacing some of the bases in the DNA with covalently labeled bases, using a series of enzymatic steps. These two methods are the most popular, as they incorporate many labels per probe, increasing the signal to noise ratio of the data, and produce long probes, allowing very stringent annealing and washing conditions which limits false positive signals.
These DNA probes can be labeled using two different approaches. The first is direct attachment of fluorophores, while the second is attaching a non-fluorescent molecule that will bind strongly to a labeled protein post-hybridization, most commonly using the biotin/streptavidin system. While direct attachment of the fluorophore is the most straight-forward, the use of a biotin tag followed by incubation with labeled streptavidin allows for signal amplification, as each streptavidin (or other labeled avidin) will likely contain several labels. Similar systems might utilize a covalently incorporated antigen with a labeled antibody, such as digoxigenin and anti-digoxigenin, but the concept of signal amplification remains the same.
Put this article into practice
Choose a free resource to help you move forward

POSTER
Histological Stains Poster

EBOOK
Guide to Special Stains for Histology

Figure 1. By labeling the probe with a small molecule like biotin and applying a labeled binding protein like streptavidin, much greater signal intensity can be achieved compared to direct labeling of the probe with fluorophore.
While this signal amplification increases the sensitivity of the method, it increases the complexity of and number of variables in an experiment. In addition, it can come at the cost of increasing background signal if the protein reagents bind/stick to other features in the sample. This isn’t to be trivialized – changes in background signal can vary widely with the source of the reagents, the lot of the reagents within one supplier, cell line, fixation and dehydration method, academic age of the scientist, phase of the moon, and state of the economy, among other things. In addition, these background issues have to be separated from the variables already involved in the hybridization of the probe.
Despite these issues, the robustness of the system and signal made enzymatically generated probes the preferred method for RNA (and DNA) FISH, with background issues worked out through trial and error, and technique knowledge retained by the rich training traditions of academic labs. (“No, you spin to the left after washes. You only spin to the right after you add the probe. Now the whole thing is ruined…”) Using these approaches, researchers have been able to see localization of RNA from transcription to maturation, and have been able to visualize increased transcription of particular genes as an increase in fluorescent signal in response to stimuli.
The Next Step In RNA FISH
However, there remained one question that the methods described so far could not answer: How many mRNAs of a particular species are present in a particular cell? This becomes complicated in FISH systems using methods that incorporate a variable number of labels per probe in addition to using secondary signal amplification system that has a variable number of fluorophores per streptavidin. This results in the amount of fluorescent signal generated per probe binding event being highly variable. Put simply, some spots are brighter than others. So now if you have a bright spot, is it one bright probe bound to one mRNA, or two or three mRNAs bound by probes of average brightness? The desire to accurately quantitate the number of RNAs in each cell has driven researchers to investigate the use of chemically-synthesized, labeled oligonucleotide probes for RNA FISH.
While oligonucleotide-based probes have defined signal per probe, these probes have two major drawbacks: very low signal per probe and potentially reduced specificity compared to the longer probes. The low signal is easily overcome by simply using a panel of oligos that all anneal to the same target, since they each have a relatively small footprint on the target.
This approach yielded some excellent results for the Kevin Fogarty and Robert Singer labs when the researchers used a handful of multiply labeled oligos per target, but the method was slow to catch on due to the difficulties (and perhaps the expense) of synthesizing and purifying oligos that contain several internal labels.
The breakthrough was to think beyond a handful of multiply labeled probes and extend the idea out to 30-48 (or more) independent, singly-labeled oligonucleotide probes per RNA species. This number of probes not only increases signal by putting more fluorescent labels on the RNA, but also because the probes bind cooperatively.
Although RNA is often drawn as a linear, extended molecule, in reality it is a highly folded structure, base pairing with itself both locally and distally to form very stable secondary and tertiary structures. This folded structure is very difficult for a short DNA oligo to bind to, and this likely frustrated earlier attempts to use oligos as probes. However, once one oligo does bind, it partially destabilizes that section of the RNA fold, allowing the next oligo to bind more easily, and the next oligo even more easily, and so on, denaturing the structure of the RNA.

Figure 2. RNA (black) can adopt a highly stable folded structure that resists hybridization with small DNA probes (red). However, with a large number of different probes complementary to the RNA, annealing becomes cooperative, resulting in well labeled RNA.
The specificity issues are a little more difficult to deal with, but can be addressed by excluding probes that bind to regions of the RNA that are of low complexity (ie. sequence regions that aren’t very discriminating to your gene of interest) and matching the annealing energies (Tm) of the various oligos used. In addition, off target interactions between one particular oligo probe and a RNA that shares some homology with the RNA of interest likely wouldn’t benefit from the cooperative binding described above.
This method was shown to be effective in a Nature Methods paper published out of Alexander van Oudenaarden’s and Sanjay Tyagi’s labs at MIT and New Jersey Medical School, respectively. In this and subsequent papers, it has been shown that this method can robustly detect single mRNA transcripts, and produces some simply stunning images. The method can be applied to both cells and tissue in a variety of species, and has been demonstrated in human, mouse, rat, yeast, Drosophila, and C. elegans.

Figure 3. Spectacular examples of RNA-FISH in action, kindly supplied by Biosearch Technologies. A: GAPDH mRNA (red) + GAPDH protein (green) in HEL-299 cells, B: Over-expressed EGFP protein (red) + GAPDH mRNA (green) in HEK-293 cells
To make things even simpler, there are now companies streamlining the whole process. They take the sequence of the RNA that you’re interested in, design a suite of matched oligonucleotide probes side-stepping any dodgy areas of the sequence likely to cause background, and then synthesize them for you, labeled with your choice of fluorophore.
Have any hints or tips for RNA FISH (or DNA FISH, for that matter)? We would love hear about them in the comments! Check out our related article for more information on didfferent FISH techniques available.
*Please don’t inject your undergraduates with purified proteins. It is unsafe, unethical, and they rarely return to the lab to let you check the antibody titers in their serum anyway.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.

Put this article into practice
Choose a free resource to help you move forward

POSTER
Immunofluorescence Troubleshooting Guide
POSTER
Histological Stains Poster