If you’re planning on using lentivirus for your next experiment, chances are you’re wondering how much virus to use. For in vitro work, multiplicity of infection (MOI) is the theoretical number of virus particles applied per target cell. That is to say, if you have 1 million cells and you want an MOI of 5, you need 5 million lentivirus particles for your transduction.
Once you have a rough idea of the number of virus particles required, you can calculate the volume and titer of virus needed to achieve your target MOI. At this stage however, researchers new to gene therapy work often run into problems. Having calculated an ideal amount of lentivirus for their experiments, they discover that the yield from their lentivirus production is too low. This usually leads to repeated attempts to produce more virus, leading to further frustration – colleagues and commercial providers routinely achieve high titers! Why can’t I?
Don’t Panic When It Goes Wrong!
Before you embark on endless rounds of optimization (a different transfection reagent, new cells, etc.), put your pipette down and listen up! It’s quite likely that a low lentivirus titer is not down to you being an incompetent lab chimp. There are several factors controlling lentivirus titer to varying degrees.
There’s no concrete limit to the number of times you can passage your cells before you see a drop in virus titer, but the cells must be healthy and actively dividing.
Clumpy cultures with lots of senescent cells won’t yield good titers.
Always do a test transfection of your cells before attempting virus production. If your transfection efficiency is poor there’s no point continuing with virus production. New cell stocks are needed!
Culture Conditions
It’s best to split your cells the day before transfection, to make sure they are happy and healthy. Optimal cell confluency at the time of transfection depends on your chosen transfection agent. Lentivirus particles are sensitive to changes in pH, so adding HEPES buffer to the culture media can protect the virus from pH extremes.
Transfection
For efficient virus production, you need to get all of your plasmids inside the maximum number of cells, so it goes without saying that you need the best possible transfection efficiency. There are many different commercial and non-commercial transfection reagents available.
Bear in mind that if you need to prep lentivirus regularly, commercial transfection kits can be an expensive option. Cheap chemical reagents like calcium phosphate and polyethylenimine (PEI) work well and are cost-effective. There’s some evidence (mostly anecdotal) that certain transfection reagents result in higher virus titers. Interpret these results with caution, as there is no real consensus on the best reagent to use. If you are new to the field, you can certainly compare transfection reagents, but don’t spend months on this step. If you are getting good cell transfection then it’s probably best to stick with what you’re using.
Plasmids
Your lentivirus plasmids don’t have to be purified using a specific method. No matter what your professor claims was the done thing back in 1998, you don’t need to use super pure endotoxin-free kits or cesium chloride! Just ensure that your plasmid is of good quality and a reasonable concentration (over 100 ng/µl). Again, it’s not worth obsessing over this step. It’s actually possible to make virus from ordinary miniprep DNA, although you may see a drop in titer.
As far as packaging plasmids go, the main rule is not to use a 3rd generation plasmid to make a 2nd generation lentivirus. Second generation transfer plasmids require the presence of the HIV-1 tat protein, which has been removed from 3rd generation systems. There’s some evidence that 2nd generation plasmids may produce higher titers when used to package a 3rd generation transfer plasmid, but the difference (if any) is probably negligible (1).
Lentivirus Transfer Plasmid
The transfer plasmid contains your transgene (or shRNA, gRNA, non-coding sequence etc.) of interest. This plasmid also contains HIV-1 long terminal repeat (LTR) sequences, essential for virus production.
Plasmid Design and Construction
Before you commence virus production, check your plasmid using sequencing and/or restriction digestion to make sure no recombination or deletions have occurred. Lentiviral plasmids are prone to mutations in some bacterial strains – use a recA- strain (e.g. Stbl3, SURE2) if possible. Trying to make virus using a damaged transfer plasmid is pretty much doomed to fail. Check out this article for tips on choosing the right bacterial strain.
When designing your transfer plasmid, do not include a polyA sequence directly after your transgene. The 3′ HIV-1 LTR already contains a polyA sequence; so inserting another one will reduce plasmid stability and virus titer.
There’s also published evidence that the use of multiple promoters in lentiviral plasmids results in ‘promoter interference’ (2). This means that one or both promoters adversely affect expression from the other. This phenomenon can decrease transgene expression and potentially virus titer.
Transfer Plasmid Effects on Virus Titer
Now that you have healthy cells, a good transfection reagent and the right packaging plasmid, you’re ready to make virus! But now we come to the most important factor in virus production: the transfer plasmid itself.
All lentiviral transfer plasmids are not created equal. One of the main factors influencing virus titer is the length of the sequence between the two HIV-1 LTRs. Yield decreases steadily and predictably as the packaging sequence gets longer. For example, if your transfer plasmid contains a short promoter driving a small transgene such as a fluorescent protein, you are quite likely to see good titers. In contrast, if you are trying to make virus using a transfer plasmid with several transgenes, multiple promoters and elements such as IRES and WPRE sequences, your virus titer will be much lower.
Different transgenes can also affect titer. Bacterial proteins such as Cas9 and Channelrhodopsin appear to be poorly tolerated by 293 producer cells, adversely affecting virus production.
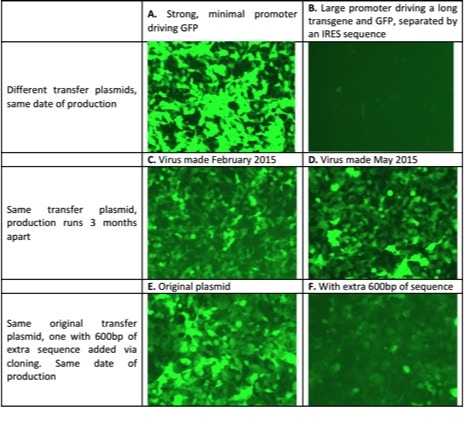
It’s difficult to overstate how important the transfer plasmid is to the success of your lentivirus production. It is not uncommon to see greater than 50-fold differences in titer between different virus batches made at the same time using the same reagents but with different transfer plasmids. Figure 1 below illustrates the variations in lentivirus titer that can occur depending on the transfer plasmid.
Figure 1: Lentivirus expressing GFP used to infect 293T cells. Fluorescence microscopy used to illustrate how various factors affect lentiviral titer and transgene expression
Therefore, promoter choice can also cause titer variations depending on how you assess transduction. Fluorescence-independent methods such as drug selection, qPCR and RT-qPCR can be used instead but they are usually more expensive and labor intensive. Photo 1B illustrates an issue that can confound your virus titer calculations. Fluorescence microscopy is a quick, simple and cheap way to measure transduction, but protein expression levels will influence your calculations. For example, fluorescent protein expression may be reduced by as much as 80% if the encoding gene is placed downstream of an IRES sequence. In this situation, you may have plenty of virus transduction but if the fluorescence signal is weak, you risk underestimating virus titer. FACS is generally more sensitive than microscopy but weak fluorescence can still cause underestimation of titer.
Going Forward
The take home message here is to avoid focusing on small tweaks that result in you spending excess time and money unnecessarily. The transfer plasmid is usually the biggest factor controlling titer so there’s not much point obsessing over optimizing steps resulting in a 10% improvement in titer if your transfer plasmid is reducing yields by 50%.
Before beginning virus production, plan your experiments and calculate how much virus you need. In vitro experiments often don’t require very concentrated virus, so there’s no point aiming for very high titers if you don’t actually need them. Bigger isn’t always better! Obviously it’s best to have good transfection levels but your cells probably won’t thrive if you bombard them with an MOI of 1000. Be realistic.
If your virus preps just aren’t producing enough for your planned experiments, consider re-cloning or redesigning your transfer plasmid. It may seem like an unnecessary detour but it could save you time, money and sanity in the end!
Most eukaryotic proteins exist as several isoforms, differing in posttranslational modifications, which allows them to perform slightly different functions or the same function under slightly different conditions. A common posttranslational modification of proteins is glycosylation.
This article is not for the die-hard old-school gel runners. You know who you are, the purists, the “I always make my own gels and buffers from scratch” kind. For you we have lots of articles about PAGE gels, both bis-tris and the standard SDS PAGE kind. Instead this article is for the rest of…
Is your goal to purify a substantial amount of a specific protein? Do you have a quantity of a molecule that binds your protein of interest? If so, generating a custom affinity matrix may be just the trick you need to purify your protein of interest by affinity chromatography. Customizing your affinity chromatography is an…
So, you’ve done your experiment, prepped your samples, and run your SDS-PAGE gel. Now it’s time for the all-important transfer step, that tricky point that will determine the quality of your Western blot. Transfer times are empirical and based on your own particular samples, which means that there is no easy way to determine how…
This is the first of a three part series describing some of the most common methods for concentrating proteins. In later installments I’ll discuss using protein precipitation and chromatography to concentrate a protein. However, here I’ll detail the most popular approach – semi-permeable membranes, used for both dialysis and commercial protein concentrators. Structure of the…
10 Things Every Molecular Biologist Should Know
The eBook with top tips from our Researcher community.