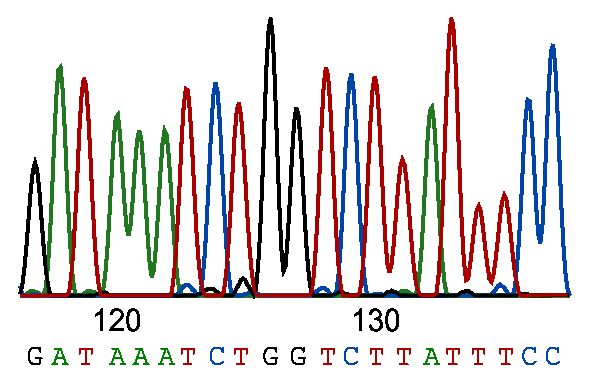
Perhaps one of the most significant discoveries in modern genetics, after the human genome was sequenced, is the role of genetic variations in evolution, disease, and the creation of plants and animals.
While the Human Genome Project (and many other genome projects, for that matter) showed how many genes living things share, they also demonstrated the importance of genetic variants.
These variants are vital to successful evolution; some genotype changes (usually of the smaller type) can lead to changes in phenotype.
But what are they, exactly, and what types of variants exist? How do they impact disease, development, survival, and medicine? And what do you need to consider when studying them? This article answers all those questions.
What Are Genetic Variants?
Genetic variants are differences in DNA sequence that occur naturally between individuals in a given population.
They appear in many forms, including single nucleotide changes (SNPs), small insertions or deletions, or larger structural changes in the DNA.
Note that these differences can occur in both coding and non-coding regions of the genome and, as mentioned, can influence various traits and characteristics, including disease risk, drug metabolism, and physical traits.
Some genetic variants are inherited from parents, while others arise spontaneously during cell division or due to environmental factors.
Key Variants Explained
Over the past 50 centuries, nearly one million variants have appeared in humans. Most of these variants are unlikely to impact our health, yet many have been linked to disease.
The search has been compared to finding a needle in a stack of needles because of the comparative rarity of genetic variations and the copious amount of genetic code that needs to be studied to confirm they exist and assess their biological significance.
We’ll discuss why genetic variants are difficult to study in more detail later. For now, here are the main types of variants that exist.
Structural Variations
This is basically an umbrella term referring to SNP/SNVs (see below), indels, copy number variations, and several other variants that change the sequence of base pairs in a genome.
These variations, while small compared to a frameshift mutation, are increasingly important in understanding human diseases. In fact, it’s been found that nearly all human tumors have some structural variants (some just a handful, others in the thousands).
Single-Nucleotide Polymorphisms/Single-Nucleotide Variations
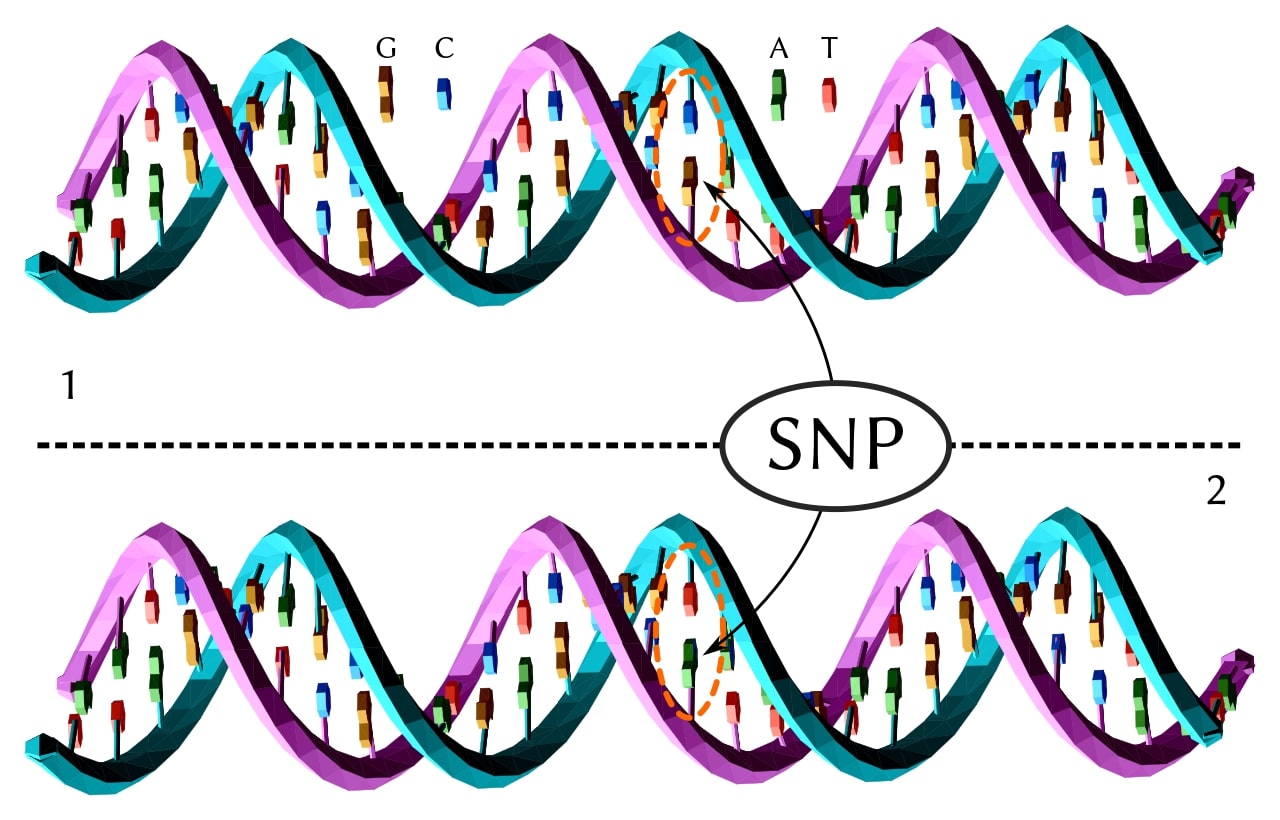
Known as single-nucleotide polymorphisms (SNPs) in populations and single-nucleotide variations (SNVs) in individuals, these variants are simply exchanges of one nucleotide base pair for another (Figure 1). There are several million SNPs in the average human, and perhaps as many in plants. [1]
These have become very important markers for certain diseases, such as breast cancer, and will undoubtedly serve as guideposts for developing personalized treatments. In fact, there are ~180 SNPs associated with the development of breast cancer. [2] Along with the known significance of mutations in the BRCA1 and BRCA2 genes, these variants are an excellent and promising example of the medical importance of genetic variations and their role in cancer diagnosis, among other therapeutic applications.
Insertion and Deletion Variations
Indels, short for “insertion” and “deletion”, are added or subtracted base pairs in a segment of DNA. It’s estimated that humans have several million of these. [3]
More substantial than SNP/SNVs, indels involve between 1 and 10,000 base pairs. Like SNP/SNVs, they most likely play some role in disease and may play an important role in determining personalized medicine. In the disease cystic fibrosis, for example, indels are responsible for the deletion of a single amino acid that triggers the disease.
Copy Number Variations
This refers to differences in the number of specific genes for a certain trait found in a genome. While the “central dogma” taught us that there were two copies of a gene in every genome., recent advances have shown that there may be many copies of a gene or none.
Copy number variations can lead to disease states. These variations may be the most prevalent of all; their large size has meant that they may involve three times as many base pairs as SNP/SNVs, the next-most prevalent structural variation.
Translocations and Inversions
These are chromosomal rearrangements of genes (or at least segments of DNA), in which the DNA segments are broken off and either located at some other point on the chromosome (translocation) or reinserted into the chromosomal DNA in “reverse,” 180 degrees from its previous alignment (inversions).
Generally, the larger the segment of DNA that is subject to these rearrangements, the more likely it will cause a change in phenotype.
Why Are Genetic Variants Difficult to Study?
Identifying genetic variants can be difficult for several reasons:
Genome Complexity
The human genome is complex. It contains around 3 billion base pairs of DNA, and the vast majority of this DNA is non-coding. Many variants can occur in non-coding regions that may not have an obvious effect on the phenotype. Good luck detecting those!
Genome Diversity
The human genome (most genomes, in fact) is also really diverse, with millions of genetic variants that differ between individuals. So distinguishing between benign variants and those that have a significant impact on health is a massive challenge. Then think about doing it at a population-wide level.
DNA Sequencing Limitations
Traditional sequencing methods like Sanger sequencing are time-consuming and expensive, given the amount of genetic data that must be sequenced. The ability to sequence large numbers of genomes quickly and accurately is limited by sequencing techniques, therefore.
Thankfully, massively parallelized next-generation sequencing methods promise to alleviate this burden.
Some Variants Are Intrinsically Difficult to Detect
Genetic variants that involve structural changes in the DNA, such as duplications, deletions, and inversions, are hard to detect using traditional current sequencing methods. A technique to detect this that’s as powerful as DNA sequencing would be a boon.
Repetitive Sequences
Repetitive sequences, of which there are loads in the human genome, are usually difficult to sequence and align to a reference genome. This can cause errors in variant calling.
False Positives and False Negatives
Sadly, not every finding we make is real! Variants can be incorrectly called due to sequencing errors, misalignments, or other technical artifacts, leading to false positives. Conversely, some true variants may be missed due to low sequencing depth or other limitations, resulting in false negatives. What a minefield.
Things to Consider When Studying Variants
Naturally, geneticists and other scientists are very interested in studying variants. And you might be interested in studying them too. So what should we keep in mind? Here are several methods and factors to consider.
Sample Size
When studying variants, the sample size is crucial to properly determine whether a variation occurs in just one genome, down to experimental error, or is a genuine and significant finding. Sample sizes needed for genome-wide studies, for example, may have to be so huge as to prohibit routine analysis.
For more information about selecting appropriate sample sizes, check out these comprehensive reviews and studies. [4,5]
Genome-Wide Association studies (GWAS) initially showed the extent of these structural variants in the human genome and others. However, GWAS or whole-genome sequencing (WGS) is still not an economical or practical way to study individual variations and groups that may be associated with the variation in a certain gene or region of DNA.
Hot Spot Analysis
For well-studied diseases such as certain cancers, some NGS manufacturers sell panels that contain sequences of known cancer genes.
For example, Ion Torrent (now part of ThermoFisher Scientific) has a “hot spot” panel that contains 207 primer pairs that match 50 oncogenes and tumor suppressor genes, including KRAS, BRAF, and EGFR. Sequencing is then carried out using any of the brands of next-generation machines (Ion PGM, Roche 454, Illumina MySeq, etc.)
This is a fast way to determine if your tumor cell samples show any variations in these genes. However, these panels cannot tell you if you’ve discovered a whole new cancer gene.
Consider Sequencing Just the Exome
When whole genome sequencing proves to be too time-consuming or expensive, sequencing the exome (coding regions) is a viable alternative option.
This technique assumes, of course, that your variants lie within genes or other coding regions of DNA.
Whole exome sequencing has been invaluable in discovering variants behind diseases, phases of development, and normal phenotypic variations. However, for epigenetic studies and searches for de-novo variants that might lie outside of coding regions, this technique isn’t helpful.
ChIP
Chromatin Immunoprecipitation (ChIP) involves pulling down cross-linked protein-DNA complexes in a chromatin preparation with an antibody directed against a certain histone modification, which can then be analyzed by qPCR or sequencing.
This technique has been useful for detecting variants that may not have been caused directly by expressing DNA (i.e., genes), showing the potential roles played by non-coding DNA and epigenetics in the development of variants.
Software for Variant Calling
Whatever the sample, and however this sample is sequenced or isolated, variants still need to be accurately called or identified.
Because the amount of data generated by whole genome sequences and even exome-specific sequences is enormous, bioinformatics techniques have been developed to determine true variants and (one hopes) weed out false positives and negatives.
There are too many software packages to list here, but for a detailed review of what’s available, be sure to read this excellent survey that summarizes it in table format. [6]
Small Alterations, Big Changes. Genetic Variants Summarized
Clearly, genetic variations are complicated. Studying them evidently provides insights into the underlying mechanisms of various biological processes and helps to identify potential targets for medical treatments.
Now you have a great overview and can start drilling deeper into specific areas.
We’ve looked at the key variations that exist and explored the link between key variations, disease, and treatment. Clearly, this still a lot to learn on this front.
Plus, if you research genetic variations, we’ve looked at several factors to keep in mind and equipped you with some helpful resources.
If you’ve found this article helpful, or if you have something to add, we’d love to hear it! Get in touch or leave a comment below.
Originally published May 2015. Revised and updated April 2023.
References
- Kumar S, et al. (2012) SNP discovery through next-generation sequencing and its applications Int J Plant Genomics. 831460
- Lilyquist J, Ruddy KJ, Vachon CM, and Couch FJ (2018) Common genetic variation and breast cancer risk-past, present, and future. Cancer Epidemiol Biomarkers Prev 27(4):380–94
- Mullaney JM, et al. (2010) Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet. 19(R2):R131–6
- Witte J (2012) Rare genetic variants and treatment response: sample size and analysis issues. Stat Med 31(25): 3041–50
- Hong EP and Park JW (2012). Sample size and statistical power calculation in genetic association studies. Genom Inform 10(2):117–22
- Pabinger S, et al. (2013) A survey of tools for variant analysis of next-generation genome sequencing data. 15:256–27