
Genomes are too large to be sequenced in one piece; they must first be chopped up into overlapping fragments, which are then reassembled based on their overlapping sequences. Sequencing a fewer number of larger fragments rather than a greater number of smaller fragments makes genome assembly easier and more reliable since each piece contains more distinctive sequences. [1] Long reads can also help find large, complicated genetic variants and can be invaluable in epidemiological studies that rely on microbial DNA fingerprints.
The two primary producers of long-read DNA sequencing technologies, Pacific Biosciences (PacBio®) and Oxford Nanopore Technologies, can routinely generate single-molecule reads hundreds of kilobases in length. [1] However, these advances have created new challenges in DNA handling and preparation. The high-molecular-weight (HMW) DNA used in long-read sequencing is more fragile than DNA used in short-read sequencing and requires unique methods for extraction and purification that minimize shearing. It is essential to ensure the integrity of the starting sample by assessing DNA quality, shearing profiles, and library size. One way to do this is to analyze HMW DNA sequencing libraries via electrophoresis. This article reviews several electrophoresis-based options to resolve and determine the size of HMW DNA.
The Importance of Quality Control in Long-Read DNA Sequencing
The small DNA molecules used in short-read sequencing (75-300 bp) are quite robust and can easily withstand extraction procedures, bead purifications, and shearing protocols. Assessing DNA quality via size analysis is generally quick and painless using a Bioanalyzer chip or TapeStation (Agilent) or merely running a midi slab gel.
In contrast, HMW DNA—fragments over 10 kb in length—can break at any one of several library construction steps. Every pipetting step (slowly, with wide-bore tips is recommended!) can break the DNA into small fragments that can affect your sequencing results. During DNA sequencing, the presence of smaller molecules in a library reduces the average read length, eliminating the primary benefit of long-read sequencing: the ability to more easily and correctly reassemble the fragments. [3,4] Shorter length DNA can be removed from libraries with methods like Sage Science’s BluePippin High-Pass DNA size selection. However, this can impact DNA yield depending on the fragment size cut-off and fragment size distribution of pre-selection library. [5]
With or without size selection, it is important to evaluate the starting DNA quality when working with HMW DNA by assessing the post-shear fragment distribution and the final library size. The following methods allow you to do just that so that you can be sure that your long-read sequencing libraries do, in fact, begin with long pieces of DNA [5,6].
Electrophoresis-Based Methods for Analyzing HMW DNA Size
As any molecular biologist can tell you, gel electrophoresis works by using an electric field to move DNA through a molecular sieve. Small molecules travel through the sieve more easily and quickly, while larger ones get tangled and move more slowly. You can determine the size of your DNA sample by comparing its position in the gel to that of a known standard, usually in the form of a size ladder. Resolving HMW fragments that exceed the size of the pores is problematic, though, as larger molecules (15-20 kb) may not move through the gel at all. Several electrophoretic methods have been developed specifically to resolve these larger DNA molecules.
Pulsed-Field Gel Electrophoresis
In the early 1980s, groundbreaking work by Shwartz and Cantor [2] showed that using an alternating, pulsed electrical field as opposed to a direct current can resolve HMW DNA up to 2000 kb. [2] In pulsed-field electrophoresis, the voltage is switched periodically among three directions instead of constantly running in one. [3] DNA molecules respond to the voltage changes by realigning their charge at different rates based on their size, with smaller pieces adjusting more quickly. Over time, even long DNA strands are propelled forward.
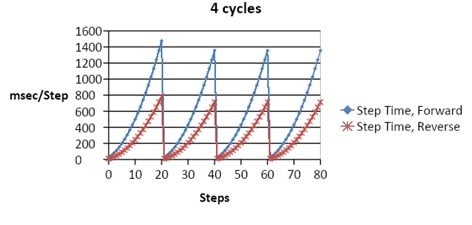
This “two steps forward, one step back” approach is an effective way of separating large pieces of DNA. However, it can be complicated, time-consuming, and requires specialized equipment. Field reversals are usually short (milliseconds to seconds) and are often modulated incrementally to achieve the desired results. For instance, users may require high separation within a fragment size range while retaining high compression within a and another range of fragments. Figure 1 provides an example of a few seconds of a pulsed-field pattern.
Femtopulse (Pulsed-Field Capillary)
Agilent’s Femtopulse is a capillary-based system that can resolve DNA from 1300 bp to 165 kb. This size range is well within the distribution of PacBio’s single-molecule sequencing libraries, which are typically between 15 kb and 100 kb. However, the 165 kb maximum limit may not provide an accurate depiction of larger fragment distributions, such as Oxford Nanopore systems.
Femptopulse is a great way to measure library size distributions and quality checking starting DNA (degraded DNA contains fragments that smear below 50 kb). Its major advantage over slab gels is its user-friendly analysis software, which quickly provides accurate sizing and quantification. It is also preferable when sample sizes are limited. The Femtopulse is expensive, so it is probably best suited to dedicated high-throughput labs where its cost can be justified (e.g., those labs using PacBio Sequel II or Oxford Nanopore PromethION).
CHEF Mapper (Pulsed-Field Gel)
The BioRad CHEF Mapper XA System is based on work by Schwartz and Cantor, [2] but allows more flexibility over the angle of the pulsed-field. It can resolve ultra-high-molecular-weight DNA fragments, from 100 bp up to 10 Mb in length. Nonlinear switch-time ramps change the switch time increments during the run and separate fragments from 50 to 700 kb. Secondary pulses can facilitate separation and resolution of larger molecules by releasing large DNA molecules stuck in the gel matrix. The CHEF Mapper is more affordable than the Femtopulse but requires third-party gel imaging software. Among the drawbacks is that it is a large apparatus compared to typical agarose gel setups, and can require water recirculation.
Pippin Pulse (Field Inversion Gel)
Sage Science Pippin Pulse is a simple option at a fraction of the cost of the Femtopulse or CHEF Mapper. It uses field inversion (back-and-forth field reversal) on a typical midi-gel size box with reinforced platinum electrodes to withstand the pulse regimens. The Pippin Pulse can resolve DNA up to 400 kb, can handle most of the chores in a PacBio or Oxford workflow, and takes up very little bench space. Gels are cast like any midi-gel, and it uses a standard power supply. When running the gel, simple software applies either preset or user-defined pulsed-field or direct-current protocols. Like CHEF Mapper, third-party imaging software is required for sizing and quantification. Figure 2 shows an example of a Pippin Pulse protocol.
Summary of Electrophoresis-Based Methods for Analyzing HMW DNA Size
The table below provides a summary of the different electrophoresis-based methods for analyzing HMW DNA size, based on cost, size, resolution, and any need for third-party software.
Method (company) | Method | Resolution | Cost | Size | 3rd party equipment/ software needed? |
(Agilent) | Capillary electrophoresis | 1300 bp to 165 kb | Highest | 51 x 61 x 35 cm | No |
(BioRad) | Pulsed-field gel electrophoresis | Up to 10 Mb | Midrange | 55.9 x 34.5 x 30 cm | Yes |
(Sage Science) | Field inversion gel electrophoresis | Up to 400 kb | Lowest | 7 x 20 x 24 cm | Yes |
You can only reap the benefits of long-read sequencing if you start with intact fragments of long DNA. These tools can help confirm that your HMW has not been sheared during isolation and purification, enabling you to get the cleanest reads possible.
For more on the upstream step that sets all this up, see why size selection is key for long-read sequencers.
References
1. Kumar KR, Cowley MJ, Davis RL. Next-Generation Sequencing and Emerging Technologies. Semin Thromb Hemost. 2019; 45(7):661-673. doi:10.1055/s-0039-1688446
2. Schwartz DC and Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37, 1. doi: 10.1016/0092-8674(84)90301-5
3. Introduction to SMRTbell™ Template Preparation. Pacific Biosciences product documentation.
4. Sequencing library preparation for MinION™ and PromethION™. Oxford Nanopore product documentation.
5 Schalamun, M. et al Harnessing the MinION: An example of how to establish long?read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora Mol Ecol Resour. 2019;19:77–89. Doi: 10.1111/1755-0998.12938
6. Blethrow, J., Best Practices for Whole Genome Sequencing Using the Sequel System. Pacific Biosciences scientific poster, 2018.
7. Pippin Pulse User Manual. Sage Science product documentation.






