
Despite the rising popularity of CRISPR/Cas genome engineering, RNA interference (RNAi) still remains a widely used tool in the life scientist’s toolbox (brush up on the mechanism of RNAi here). In fact, RNAi recently entered the clinic with the approval of Patisiran, an siRNA-based therapeutic for a rare form of neuropathy related to hereditary transthyretin amyloidosis. [1] Such a feat would have been considerably more difficult, if not impossible, if not for the discovery and optimization of chemical modifications that improve siRNA efficacy. Chemical modifications have been increasingly favored by researchers to improve the nuclease-resistance ability and delivery of siRNAs while mitigating their immunogenicity (i.e., their tendency to provoke an immune response).
To illustrate the types of chemical modifications that can be made to siRNAs, let’s first take a look at the basic structure of RNA. Figure 1 depicts two ribose nucleosides linked together by a phosphodiester bond. Shown in color are the sites that can be chemically modified. These include the non-ester groups of the phosphodiester backbone (orange), bases (magenta), sugars (green), and 5’/3’-covalent modifications (X, blue). These are the five ways to chemically modify siRNAs (the last category is counted twice to account for different types of conjugations).
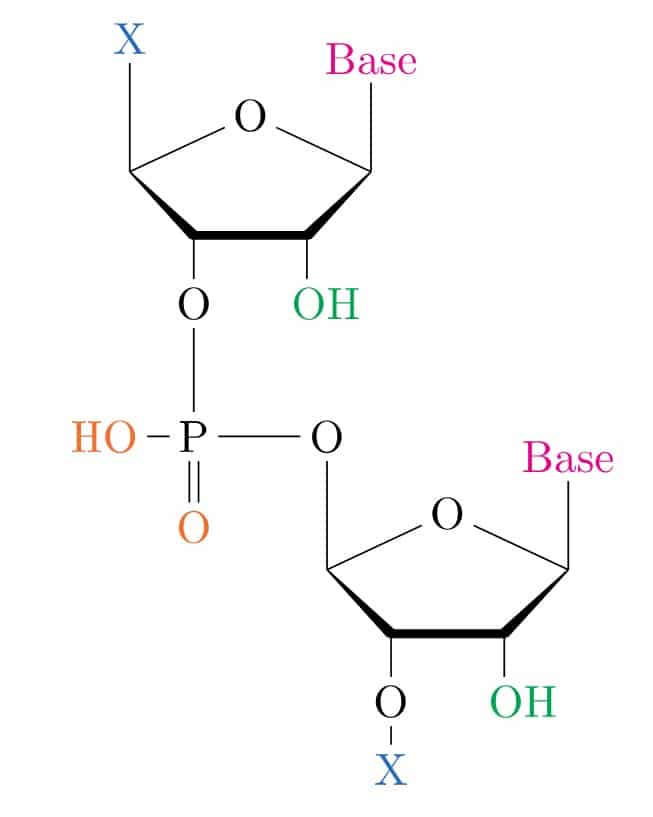
1. Termini Modification
The most common way to chemically modify siRNA is by phosphorylating the 5’ end of the antisense strand. siRNAs lacking 5’ phosphate groups at this site are not recognized by Argonaute 2 (Ago2), a key component of the RISC (RNA-induced silencing complex) machinery that mediates RNAi. However, if you don’t phosphorylate this key site yourself, don’t fret; the cell will do it for you. [2] Note though that, in general, heavily modified siRNAs can make poor substrates for intracellular kinases, [3] an obstacle that can be overcome by using 5’-phosphate analogs that cannot be hydrolyzed, such as 5’-(E)-vinyl-phosphonate. [4]
2. Backbone Modifications
Modifying the phosphodiester group that links consecutive ribose nucleosides has been shown to enhance the stability and in vivo bioavailability of siRNAs [4]. The non-ester groups (-OH, =O; colored orange in Figure 1) of the phosphodiester linkage can be replaced with sulfur, boron, or acetate to give phosphorothioate, boranophosphate, and phosphonoacetate linkages.
Choose a free resource to help you move forward
CHEAT SHEET
Nuclear Extraction Protocol
DOWNLOAD
Nanodrop Results Interpretation Card
Phosphorothioate, boranophosphate, and phosphonoacetate linkages stabilize siRNAs because they are less susceptible to exo- and endonuclease hydrolysis. However, phosphorothioate and phosphonoacetate linkages need to be used with caution as they can weaken the potency of siRNAs by inhibiting their loading into the RISC machinery. [4] They can also make siRNAs more likely to bind immune receptors, making them more immunogenic. [5] Boranophosphate linkages, on the other hand, are simply difficult to synthesize by chemical methods. [4]
Substituting the phosphodiester group with a phosphotriester facilitates cellular uptake of siRNAs and retention on serum components by eliminating their negative charge. [6] This is particularly useful for in vivo siRNA delivery, as it increases the half-life of siRNAs in circulation. Also, this modification effectively transforms small interfering ribonucleic acids [siRNAs] into small interfering ribonucleic neutrals [siRNNs] because the incorporation of the third ester removes the negative charge of the linkage. Once inside a cell, esterases remove the third ester, converting siRNNs back into siRNAs. [7]
3. Sugar Modifications
When ribose sugars are deprotonated (reaction catalyzed by exo- and endonucleases), the 2’-hydroxyl (2’-OH) group (colored green in Figure 1) can act as a nucleophile and attack the adjacent phosphorous in the phosphodiester bond. This is why modifying the phosphodiester bond improves the stability of siRNAs — by protecting them from nucleophilic attack by adjacent, deprotonated sugars.
Rather than modifying the phosphodiester bond to make it less susceptible to nucleophilic attack, the 2’-hydroxyl group of the ribose sugar can be defanged by replacing it with something less nucleophilic (i.e., less likely to donate electrons). Such alternatives include 2’-O-methyl, 2’-O-methoxyethyl, and 2’-fluoro modifications. As an added benefit, these modifications also impact the conformation of the ribose sugar due to their electron-withdrawing properties, an effect that can increase the binding affinity of the siRNA duplex. [4] Bridging the 2’ and 4’ positions of the furanose ring — called a locked nucleic acid — exhibit similar effects on siRNA binding affinity. However, incorporating too many such modifications can inhibit the dissociation of the siRNA duplex during RISC loading. [4]
Overall, sugar modifications are more common than backbone modifications because they do not impact siRNA recognition by RISC. [8]
4. Base Modification
Immune cells can distinguish foreign RNAs from ‘self’ RNAs because the former lack chemical modifications that naturally occur in mammalian cells. [4] These include sugar modifications (colored magenta in Figure 1) such as 2’-O-methyl and base substitution with modified bases such as pseudouridine, 5’-methylcytidine, N6-methyladenosine, inosine, and N7-methylguanosine. Therefore, modifying siRNAs at these sites will allow them to escape immune detection, [9] but can also reduce their silencing potency. [10]
5. Lipid Conjugation
Less common than other modifications, lipids can be conjugated to the 5’ or 3’ termini of siRNA to improve their in vivo bioavailability by allowing them to associate with serum lipoproteins. For example, cholesterol and vitamin E conjugates dramatically enhance siRNA uptake in the liver and other tissues upon intravenous injection. [4] Other fatty acids — namely, palmitate and tocopherol — have proven less toxic in mice while maintaining gene silencing activity. [11]
Should I Use Modified siRNAs?
Although not required for many in vitro applications, chemically modified siRNAs generally perform better than their unmodified counterparts. In my experience, the enhanced stability of chemically modified siRNAs effectively reduces the concentration of siRNAs needed to achieve sufficient gene knockdown. This is important because high concentrations of siRNA can lead to off-target effects [12] (Generally-speaking, you should only use as much siRNA as necessary to sufficiently silence your target). Most in vivo applications, on the other hand, absolutely require chemically modified siRNAs due to the challenges associated with in vivo RNAi (e.g., bioavailability and cellular uptake).
Thankfully, many commercial vendors — ThermoFisher, IDT, Millipore-Sigma, and Horizon Discovery, to name a few — take the guesswork out of siRNA optimization by offering chemically modified siRNAs optimized for stability and potency. You could synthesize your own modified siRNAs, but bear in mind that this may require extensive optimization at every step of the process.
Regardless of whether you obtain your siRNAs commercially or synthesize them yourself, chemical modifications must be balanced so as not to reduce gene silencing potency — after all, a highly stable siRNA is only good if it can silence its intended target.
References
- A triumph of perseverance over interference. Nat Biotechnol. 2018;36(9):775.
- Weitzer S, et al. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature. 2007;447(7141):222-6.
- Haraszti RA, et al. 5-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017;45(13):7581-92.
- Lennox KA, et al. Chemical Modifications in RNA Interference and CRISPR/Cas Genome Editing Reagents. Methods Mol Biol. 2020;2115:23-55.
- Yanai H, et al. Suppression of immune responses by nonimmunogenic oligodeoxynucleotides with high affinity for high-mobility group box proteins (HMGBs). Proc Natl Acad Sci U S A. 2011;108(28):11542-7.
- Meade BR, et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat Biotechnol. 2014;32(12):1256-61.
- Hamil AS, et al. Synthesis and Conjugation of Small Interfering Ribonucleic Neutral SiRNNs. Methods Mol Biol. 2016;1364:1-9.
- Chiu YL, et al. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9(9):1034-48.
- Kariko K, et al. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165-75.
- Sipa K, et al. Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. RNA. 2007;13(8):1301-16.
- Ostergaard ME, et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019;47(12):6045-58.
- Caffrey DR, et al. siRNA off-target effects can be reduced at concentrations that match their individual potency. PLoS One. 2011;6(7):e21503.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.





