

Why do individuals respond differently to a drug or life event? Answering this question is a crucial step in the drug or vaccine development process, and one place to start is to determine if differential gene expression plays a role.
Historically, if you wanted to study gene expression, you would use qPCR. But this approach limits the number of genes you can examine and requires prior knowledge of gene sequences, meaning identification and profiling of novel genes is not possible.
One solution is to utilize next-generation sequencing (NGS), which can facilitate higher throughput. NGS allows you to perform gene expression discovery, without prior assumptions, in a wide range of settings and identify specific genes that respond to a particular stimulus, such as an anti-cancer drug.
Whole-transcriptome RNA sequencing provides base-pair resolution for every transcript, which can be useful if you need to study mutations or splice variants. However, if your research goal is to measure the expression levels of genes, using whole-transcriptome RNA will give you much more data than you need. Excess data means spending more money and time on performing the experiments and analyzing the data than is desired.
If you are looking for an NGS method to profile gene expression that won’t blow your budget, consider 3? mRNA-Seq. This method boasts sensitive detection of gene expression, more straightforward data analysis and can deliver cost savings compared to whole-transcriptome RNA sequencing, as this article will explain.
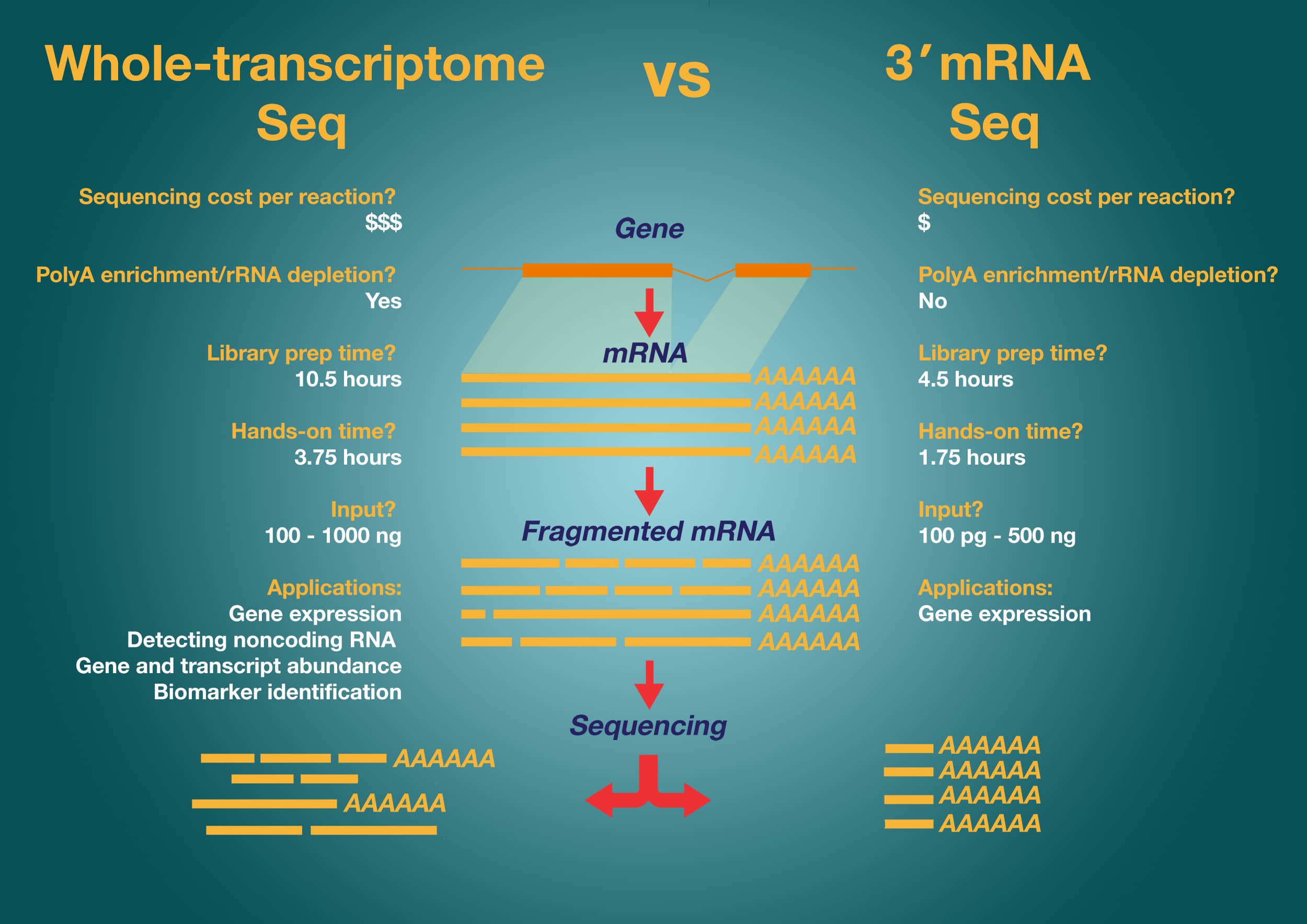
Comparison of Gene Expression Analysis Methods
To understand how 3? mRNA-Seq differs from other NGS methods of gene expression analysis, let’s compare their workflows and outputs.
Whole-Transcriptome RNA Sequencing for Expression Profiling
Whole-transcriptome RNA sequencing can detect unknown transcripts and enable the study of coding and noncoding genes simultaneously. Whole-transcriptome RNA sequencing provides you with the ability to identify previously unknown drug targets and effects and identify individuals who will respond differently to a particular treatment. Kits such as the CollibriTM Stranded RNA-Seq kit for IlluminaTM Systems make this task relatively simple. The workflow involves:
- Capturing the mRNA using magnetic oligo-dT beads
- Fragmentation of mRNA
- Addition of helper adaptors to the RNA, which form full-length sequencing adaptors for Illumina systems during PCR
- Generation of double-stranded cDNA
- Library amplification
- Library quantification
- Sequencing.
You can measure differential gene expression by comparing read count data from different experimental groups. For example, a drug-treated sample compared to a control. Differential gene expression results from whole-transcriptome sequencing correspond well to qPCR data and show high levels of reproducibility between replicate samples.
One caveat with this approach is that it requires more reads per sample compared to 3? mRNA sequencing, and so is more challenging to perform data analysis and requires a more significant investment in sequencing chemistry.
3? mRNA-Seq: A Simpler Alternative for NGS Expression Profiling
3? mRNA-Seq also offers the ability to study expression levels of many genes simultaneously and without prior knowledge of the gene sequence. As with the traditional mRNA-Seq approach, it also has high levels of reproducibility between replicate samples and corresponds well to qPCR data.
Advantages of 3? mRNA-Seq
The 3? mRNA-Seq method is similar to the traditional mRNA-Seq approach but has some key differences.
- Reverse transcription takes place only at the 3? polyA tail of the mRNA using an oligo dT primer
- The mRNA is degraded before second-strand synthesis.
These differences mean that 3? mRNA-Seq does not suffer from the same issue of bias towards longer transcripts as whole-transcriptome sequencing since it generates only one fragment per transcript. The generation of only one fragment per transcript also simplifies data analysis because there is no need to adjust for the transcript length.
Use of 3? mRNA-Seq in Gene Expression Analysis
If you’re not sure about testing out this new technology, be assured that 3? mRNA-Seq is already in use for a wide variety of research interests and in a range of different tissues and experimental conditions.
Using 3? mRNA-Seq, researchers in Austria investigated the pathways involved in immunoediting in cancer. [1] They compared the response of wild-type and immunodeficient mice to injected MC38 colon adenocarcinoma cells. From this, they were able to identify several genes linked to the immune defense mechanism triggered by the tumor, furthering our understanding of the pathways involved in this vital process.
Gastroenterology is another area that has already benefited from the 3? mRNA-Seq technology, with researchers in Germany using it to study the effect of the mouse intestinal biome on inflammation response. [2] Comparison of germ-free mice and healthy mice colonized with pathogen-free microbiota, resulted in the identification of differentially expressed genes related to macrophage biology in the germ-free mice.
3? mRNA-Seq has commercial applications too, with researchers in Thailand using it to study metabolic response in the soil fungus Aspergillus terreus. [3] This fungus produces itaconic acid, an important chemical used in many industrial processes. The researchers identified upregulated genes under conditions of increased itaconic acid production. Identification of these genes helped clarify the pathway by which A. terreus forms itaconic acid, and suggested potential future gene editing targets to optimize production.
Summing up the Benefits of 3? mRNA-Seq
3? mRNA-Seq is already being used to study differential gene expression in a variety of tissues and species and has allowed researchers to identify new components of pathways in a range of processes. This technique is furthering our knowledge on a range of topics from health issues, including cancer and IBS, to commercial interests such as optimizing the production of commercially valuable bioproducts.
When it comes to detecting the differential expression of unknown genes, 3? mRNA-Seq is cost-effective, easy to analyze and is as reproducible as whole-transcriptome sequencing. There are no additional costs for sequencing chemistry to generate a higher number of reads, and the method is compatible with all Illumina NGS systems.
References
- Efremova, M., Rieder, D., Klepsch, et al. (2018). Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nature Communications, 9(1), 1–13. DOI: 10.1038/s41467-017-02424-0
- Schmidt, F., Dahlke, K., Batra, et al. (2019). Microbial colonization in adulthood shapes the intestinal macrophage compartment. Journal of Crohn’s and Colitis, 13(9), 1173–1185. DOI: 10.1093/ecco-jcc/jjz036
- Songserm, P., Karnchanatat, A., Thitiprasert, S., et al. (2018). Metabolic responses of Aspergillus terreus under low dissolved oxygen and pH levels. Annals of Microbiology, 68(4), 195–205. DOI: DOI: 10.1007/s13213-018-1330-6


 4")

