
Even though our knowledge about genomes grows daily, and in huge leaps, we sometimes need to remind ourselves that DNA was first isolated in 1869 and its molecular structure was only identified in 1953. The PCR reaction only hit the scientific community as recently as 1983! So even though we are growing fast, we are still babies in the genetics field. And let me tell you, you will feel this fact intensely when you are analyzing a whole genome (or even an exome)! Classifying gene variants is one particularly challenging area of genomics. As the name suggests, gene variants are alterations to a given DNA sequence. Gene variants can be benign, pathogenic, or of unknown significance.
Why is it important to study and classify these variants? Well, while many gene variants don’t translate into disease, or susceptibility to disease, some are associated with serious diseases, such as various forms of cancer, hemophilia, neurofibromatosis, and Progeria (a disease characterized by accelerated aging). Thus, classifying gene variants is extremely important, because knowing their physiological consequences may help to distinguish between healthy individuals and those susceptible to certain diseases e.g., cancers. This could contribute to screening programs, preemptive medicine, and better patient outcomes.
Why Is It So Hard to Classify Gene Variants?
Well, there are many reasons. But the real enemy here is ignorance. And the real problem? The sheer volume of information we each store in our DNA:
- Humans have 46 chromosomes, and are diploid organisms. This means that we have 23 pairs of chromosome. We can think of it as 23 chromosomes inherited from our mother, and 23 chromosomes inherited from our father.
- It is estimated that humans have around 20,000 protein-coding genes (however this number is evolving, as new information emerges each day!)
- 98% of the genome is comprised of non-coding DNA that does not encode protein. The fact that it doesn’t translate into an amino acid sequence doesn’t make it irrelevant! The regulatory sequences that control basic cellular functions of the cell are located in the non-coding DNA. Variants in these sequences could greatly affect the homeostasis of the organism.
- Our genome size is 6,469.66 mega base pairs. That is a lot of information stored in our DNA!
It’s already looking like a tough job to classify a gene variant. However, the job gets even more difficult. There are small differences between individuals, and therefore everyone’s genome is slightly different! So, you have to be able to distinguish between low-represented polymorphisms and a gene variant that could cause disease.
Enjoying this article? Get hard-won lab wisdom like this delivered to your inbox 3x a week.

Join over 65,000 fellow researchers saving time, reducing stress, and seeing their experiments succeed. Unsubscribe anytime.
Next issue goes out tomorrow; don’t miss it.
Indeed, it’s like trying to find a particular needle in hundreds, and hundreds of haystacks, filled with different needles – that you will have to analyze in depth to make sure it is not your needle. It may seem like I am exaggerating, but trust me, I am actually making light of this subject.
How Can We Make It Easier to Study Gene Variants?
Thankfully, there is a great community of scientists and researchers who dedicate their careers to make our gene variant hunting journey easier. Hundreds of human genomes have been sequenced, giving us something to compare our results to. There are many public databases with gene variants known to be associated with many diseases. There are also several functional studies to try to decipher the consequences of certain gene variants on the resulting proteins. These functional studies try to shed a light into the function of the protein, but more importantly, how (and if) the protein’s function is affected by the found variant. Keeping in mind that proteins that do not function properly will probably lead to disease, these studies are very important!
The Ensembl Database
Let’s have a look at my favorite tool for studying gene variants, Ensembl.
Ensembl is a great way to start your journey into gene variant analysis. This database gives you access to thousands of genes from many species. You can just enter the name of the gene, phenotype, or any other nomenclature in the areas signaled by the arrows and you are in! (Figure 1).
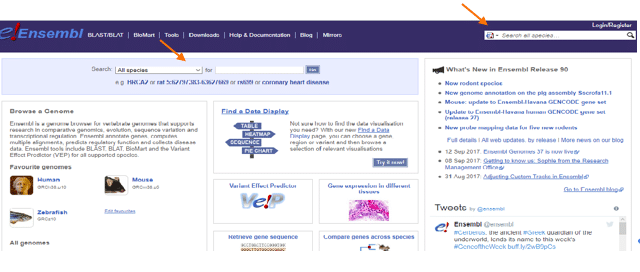
The beauty of Ensembl is that it will give you a lot of information about your gene or phenotype of interest, as well as providing links to some other amazing resources, like UniProt and NCBI.
After you search for your favorite gene, you will get an output page similar to Figure 2. Here, you will find a description of your gene, its synonyms (2A), location on the genome (2B), and other important information, including associated phenotypes (2C).

Transcript Information in Ensembl
You will also see a transcript table (2B), where you will find transcripts for the gene of interest. In this table you will find protein-coding transcripts, noncoding transcripts and splice gene variants. On the Biotype column, you will find a color-scheme categorization. The transcripts both in gold and in Consensus CoDing Sequence (CCDS) are reviewed transcripts and of high quality – this means that this transcript is equal between the automated annotation pipeline from Ensembl and the manual curation from the Vega/Havana project. The blue transcript is a non-coding transcript, while the red transcript is either from automatic annotation pipeline, directly form Ensembl, or is manual curated (Vega/Havana project).
More importantly, it informs you about which transcripts are protein-coding, and provides their NCBI reference sequences. In the example shown with the BRCA2 gene, the NCBI reference sequences (in the RefSeq column) are NM_000059 and NP_000050. NM_ refers to the mRNA, while the NP_ refers to the protein, and if you click on the hyperlinks, you will be taken to the NCBI Nucleotide database for that specific mRNA/protein. Here, you will find the FASTA sequence, literature mentioning the sequence, exons, and authors. You should always keep in mind both of these resources .
On the column on the left (Figure 2C) you will have a number of options, leading you to precious information, such as: sequence, comparative genomics, associated phenotypes (including known gene variants).
Links to Other Databases
The transcript table also gives you a hyperlinks to the UniProt database and the protein’s UniProt code. In the example shown, P51587 is the UniProt code for the first BRCA2 transcript listed in the table.
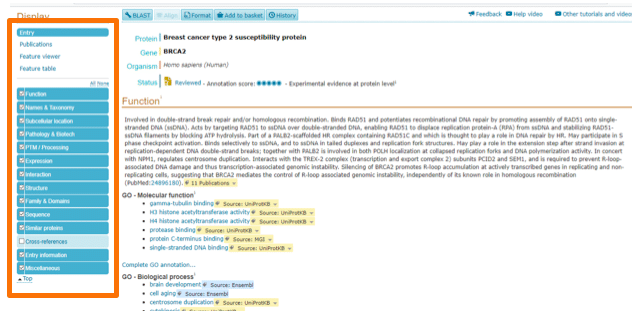
UniProt is a useful database that provides information on your protein of interest, such as: Function, Names and Taxonomy, Subcellular Location, Pathology and Biotech, and Expression (tissue specificity, gene expression databases, organism specific databases). On the Pathology and Biotech tabs you will be able to check gene variants and the diseases these have been associated with, relevant original literature and the graphical view (the localization of the variant on the gene).
In Figure 3 you can observe the UniProt page for BRCA2. On the left column you will find all of these categories (and then more), and I advise you to check all of them – you will find a lot of important information.
It’s clear that I think highly of Ensembl, and I advise you check it out in detail for yourself. You can also check these tutorials to learn more about the database. Spend some time on it to really understand the depth of knowledge available, and see it as not only the first step to classifying a gene variant, but also an important way of collecting information on your genes and proteins of interest, to hopefully accelerate your research.
Now you must be thinking: well, but how do I classify gene variants? Well, with a firm grasp on your gene, its related phenotypes, and all the nomenclature associated with it, you are ready to dive into the world of classifying gene variants. There are many tools out there, and next time we will be discussing dbSNP and ClinVar. Stay tuned for part 2 in this series!
References
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.






