Regular readers will know about the advantages of T4 DNA polymerase-mediated ligation independent cloning. The fact that it is faster, more efficient and allows easier parallel cloning than conventional cloning has made it my method of choice in the lab.
But the technique does have it’s downsides – not least the requirement that existing vector multiple cloning sites be modified to convert them into ligation independent cloning vectors.
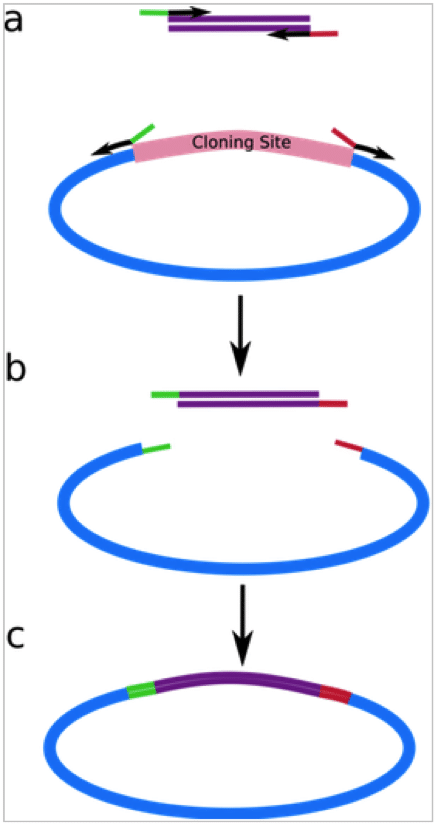
This paper by Li and Elledge recently flagged up in a comment by Max (thanks Max!) looks like it could change all that. It turns out that no sequence modification is required at all and LIC (or sLIC – sequence and ligation independent cloning as the authors call it) can be performed at any site in any vector of your choice.
If you are familiar with T4-mediated ligation independent cloning you will know that the vector sequence needs a specific LIC site containing restriction site flanked by regions that lack one of the nucleotides (e.g. adenine) for a 13-14nt stretch (read this first if you are not familiar with it). After linearising at the restriction site, the vector is incubated with dATP + T4 DNA polymerase, which chews back the 3′ end of the DNA until it stops after 13-14 nucleotides due to the presence of the dATP. This creates a single-stranded region to with a similarly treated insert can be annealed.
In this paper, Li and Elledge showed that the specific vector LIC site was not required. Treating the vector with T4 DNA polymerase and no dNTPs for a certain length of time (30 minutes was optimal for them) was sufficient for vector preparation. The single stranded stretch this creates is longer than required for annealing an insert, but single stranded gaps like these are apparently repaired very efficiently by E.coli after transformation so this is not a problem.
They also demonstrated that inserts prepared in one of three ways could be successfully annealed and transformed, which considerably increases the versatility of the process. The insert prep methods were:
1. T4 DNA polymerase treatment. Just like the vector, the insert could be subjected to T4 DNA polymerase treatment (without dNTPs) to create single stranded regions that will anneal to the prepared insert.
2. iPCR. Non-treated PCR fragments could also be annealed to prepared vectors, albeit with much lower efficiency. They showed that this is because a subset of fragments were synthesised incompletely, resulting in 5′ overhangs. Although this is was relatively in efficient, the authors found that it was robust and recommend it for routine cloning.
3. Mixed PCR. This was the most efficient method. It involves amplifying the insert using two separate reactions. In the first reaction, the forward primer has a 30nt tail homologous to the vector ss region, while in the second reaction, the reverse primer has the homologous tail. After amplification the two reactions are mixed, denatured and annealed to yield a subset of inserts that have both the forward and reverse primer single stranded tails that can be annealled into the prepared vector.
The authors showed that efficient annealing needed only 20-30 nt (single stranded) regions of homology at each end of the vector and insert. Amazingly, the homologous regions didn’t even need to be at the ends of the insert – the authors showed that non-homologous regions of up to 20 nucleotides could be tolerated as the branched products produced after annealing are efficiently trimmed and repaired by the cell.
I have not had a chance to try out this method yet, but it certainly looks very exciting. It looks to me like once this I have this protocol is up and running there will be no need at all for restriction enzyme-mediated cloning… which is a day I long for!
When I have some results of my own from this I’ll publish them, and my protocol here so watch this space.