
Manipulating the genes of organisms is crucial for studying their functions. In times before genetic engineering, scientists would shoot bacteria with X-rays or expose them to destructive chemicals until spontaneous mutations would arise. Fortunately, current methods are more sophisticated and less torturous.
Researchers now use more directed techniques to introduce mutations. There are several ways to manipulate the bacterial genome and every microbiologist has their own preferred method based on a variety of reasons, including species of bacteria, resources available, and personal laboratory training.
Here, I’ll outline how to deploy a suicide vector to conduct a two-step allelic recombination.
Why Should You “Two-Step” in the First Place?
Before we go on, let’s be sure that we are all on the same page with some key words that describe ways DNA can move between organisms: conjugation, transformation, and transduction. Conjugation is a microbial mating process where one bacterium expressing an F-pilus (a name I have always found hilarious because I’m an enormous child in a lab coat) injects plasmid DNA into its bacterial neighbor. Transformation occurs when cells “take up” DNA from the environment, and transduction is the result of infection with bacteriophage DNA.
Choose a free resource to help you move forward
CHEAT SHEET
Nuclear Extraction Protocol
PROTOCOL
Chemically Competent Cells Protocol
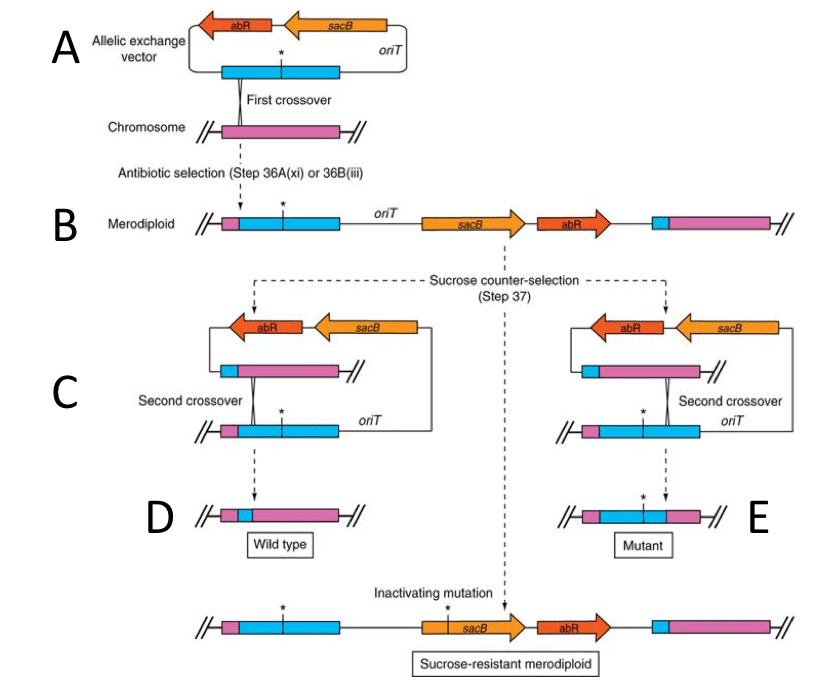
Now, back to two-step allelic recombination. Sometimes, expressing proteins on a plasmid just doesn’t cut it. If you need to knock out a gene or work with endogenous gene expression levels, directly manipulating the chromosome is the best way to go. The idea behind suicide vectors is to use a piece of DNA that cannot replicate in the host of choice. Typically, this includes a plasmid that is maintained in a compatible host but upon conjugation into the relevant species, the origin of replication is unrecognizable and, understandably, can’t replicate. This situation forces a single homologous recombination event to occur, especially under antibiotic selection. This is because the only way for the bacteria to survive in the presence of an antibiotic, for which resistance is encoded on a suicide vector, is to homologously recombine the entire plasmid into its own genome at the site that matches a patch of DNA on the vector. This is the first step of the two-step allelic exchange. A second homologous recombination event removes the integrated vector and leaves behind either the wild-type or mutant copy of the gene (Figure 1).
How to Choose and Construct Your “Suicide Squad”
Choice of suicide vector should first be determined by selecting an incompatible origin of replication. I worked with Vibrio cholerae in grad school and utilized the pKAS32 vector for chromosomal alterations. Suicide vectors typically contain multiple cloning sites (MCS) with various restriction enzyme sites to choose from. The pair that always worked for my lab was AvrII and KpnI. I would conduct SOE (splicing by overlap extension) PCR to produce an insert for pKAS with a 5’ AvrII site and a 3’ KpnI site with 1 kb of homology on either side of my gene of interest.

For example, if I wanted to delete a gene, I would PCR 1 kb fragments upstream (PCR #1) and downstream (PCR #2) of my gene of interest and then PCR those two fragments together for a clean deletion in the SOE process (PCR #3). The protocol would look something like this:
PCR #1
Forward primer: 5’ AvrII site and homology to 1 kb upstream of the start codon
Reverse primer: 5’ 20-25 bp overlap with the reverse sequence just downstream of the stop codon and a 3’ sequence matching the reverse sequence just upstream of start codon.
Template: V. cholerae genomic DNA
Product: 1 kb fragment
PCR #2
Forward primer: almost the exact reverse complement of PCR #1 R primer
– you may have to play around with the nucleotides to make ensure melting temperature of the annealing bit stays around 60oC and the overlap bit still has 20-25 base pairs
Reverse primer: 5’ KpnI site and homology to the reverse strand 1 kb downstream on the stop codon.
Template: V. cholerae genomic DNA
Product: 1 kb fragment
PCR #3
Forward primer: PCR #1 F
Reverse primer: PCR #2 R
Template: 80 ng each of PCR products from first two PCR reactions
Product: 2 kb fragment
The above protocol can be adapted to insert DNA as well. As long as primers contain overlapping sequences, the resulting products can be “SOE-ed” together using the products themselves as a template in the subsequent PCR reaction.
PCR products from each step should be gel purified to verify size. SOE product and desired vector should each be digested in a double enzyme digest overnight at 37oC. The insert is then ligated into the vector overnight at 16oC. The resulting ligation is transformed into an intermediate E. coli host.
Pro Tip: I usually digest a pKAS32 vector already containing an insert. That way, even if the ligation didn’t stick (get it?), I can at least verify the digestion worked by visualizing the already-there insert on a gel.
First Cross-Over
Once created, the suicide vector is conjugated into the bacterial species of interest (Table 1 and Figure 1A). Exconjugants are selected by the resistance cassette conferred by the vector and another antibiotic to kill E. coli parent. In my case, the only way for V. cholerae to survive in the presence of ampicillin, for which resistance is encoded on pKAS32, is to homologously recombine pKAS32 in its entirety into the V. cholerae genome at the site that matches the insert that I cloned (Figure 1B). Simultaneously, E. coli containing the plasmid conferring the ampicillin resistant gene (AmpR) is killed by polymixin B (PB) to which V. cholerae is naturally resistant. Single colonies are then picked and re-streaked onto LB Amp PB plates to verify their resistance.
Second Cross-Over
Once resistance to the antibiotic is confirmed, the antibiotics are removed and the second stressor is added. Classic counter selection methods utilize sacB or rpsL. sacB, the counter selection gene used in Figure 1, is a gene isolated from B. subtilis that confers sucrose sensitivity in gram-negative bacteria, as well as Mycobacterium (for some reason). rpsL is a ribosomal variant that confers streptomycin sensitivity. When AmpR V. cholerae exconjugants are plated on LB containing streptomycin, they grow very slowly because they express the dominantly sensitive RpsL protein. In this method, the bacterial species of choice must be naturally resistant to streptomycin. In a second, spontaneous homologous recombination event, the vector is looped out of the genome (Figure 1C). This second cross-over event removes the sensitivity to streptomycin and the colonies exhibit wild-type growth. In this selection step, the desired colonies will be large compared to their slow-growing, vector-integrated companions. A similar method is used for sacB but, instead of streptomycin, the exconjugants are plated on LB 10% sucrose.
Pro Tip: sometimes the mutant itself confers a growth defect or phenotypic abnormality. Make sure to select “larger” colonies of all types because sometimes the big, healthy looking ones have reverted to wild-type.
The Big Gamble
Our odds are typically 50:50 whether the second cross-over event occurs at the wild-type (Figure 1D) or mutant locus (Figure 1E). The most common way to differentiate successful mutants is to conduct colony PCR using PCR #1 F and PCR #2 R. In a gene deletion, the PCR product should be smaller than wild-type (in my case exactly 2 kb). In an insertion, the PCR product should obviously be larger than wild-type. If a point mutation is introduced, the PCR products will have to be sequenced to identify correct clones. If the purpose of the two-step allele recombination is to tag a gene, then a Western blot of cell lysate or colony fluorescence can differentiate the successful clones.
Recap
- Mate suicide vector into bacteria of choice.
- Re-streak onto same media that kills E. coli donor and selects for integrated vector.
- Streak onto either sucrose or streptomycin for sacB or rspL, respectively (will be sensitive and grow slowly if vector is integrated).
- Pick fast-growing, large colonies suggesting vector has been looped out.
- Run PCR products on a gel or sequence to verify the mutant locus was left behind.
And there you have it. May the cloning gods smile upon your efforts.
References:
Hmelo et al. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat protoc. 10(11):1820-1841.
Klose KE and JJ Mekalanos. 1997. Differential regulation of multiple flagellins in Vibrio cholerae. J Bacteriol. 180(2):303-316.
Link A, Phillips D and GM Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type E. coli: application to open reading frame characterization. J Bacteriol. 179(20): 6228-6237.
Skorupski K and PK Taylor. 1996. Positive selection vectors for allelic exchange. Gene. 22(1):47-52.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.





powder Dry 7")

