When I was being trained in microbiology as an undergrad, one of the first skills I acquired was the ability to quickly compare and visualize amino acid sequences using BLAST and ClustalW. 15 years later, those two programs have done nothing but improve by expanding the data contained in these databases and simplifying the user interface. Dimitris Skliros put together a great article on the BLAST tool that explains the inner machinations and how the system works.
Here, I hope to illustrate how to use BLAST in combination with ClustalW to answer some very practical questions about protein sequences that you may find yourself stumbling into as you learn to use these tools.
Got an Amino Acid Sequence? BLAST it
There are two versions of BLAST software you can use. Dimitris talked about the first, found on the NCBI website, in the aforementioned article. It contains a few more options and variables. For the sake of consistency, I will be using the BLAST tool found on the ExPASy website.
Once you open the site, you can easily address the aforementioned question of “where did this sequence come from?” Simply copy and paste your amino acid sequence into the window and click “Run BLAST.”
Upon completion, you encounter a colored, graphical representation of the similarity with different proteins identified from the BLAST database. A color scale of green to red indicates a greater and lesser similarity. It also shows areas of significant differences.
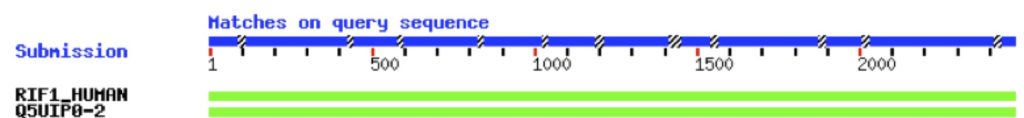
In the example shown in Figure 1, I ran a BLAST query on an “unknown” sequence and am showing the first two returned values as an illustration. They are both green, reflecting a high level of homology. You’ll notice two different naming schema in this figure. The first refers to the canonical isoform of RIF1 – hence “RIF1_HUMAN”. The second refers to “isoform 2” of this protein and uses the accession number instead of the protein name – “Q5UIP0-2”. Also, as the program considers less-similar proteins, these bars will become increasingly red, and the bar will become increasing broken, indicating gaps in the sequence.
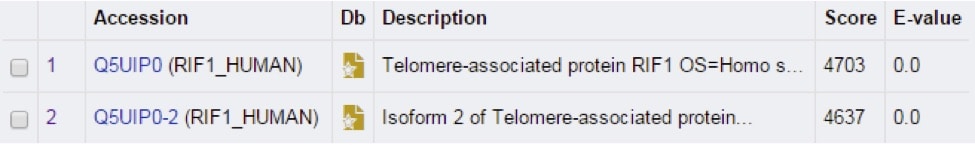
Scroll down further to see the “list of the matches.” For now, we’ll just focus on the top 2 hits again (Figure 2). Here, both the accession number, the gene name associated with this protein, and the species associated with the protein are listed. The most important values are the “score” and the “E-value” numbers. A higher score indicates a better match between the two sequences. The E-value measures biological relevance – the lower the E-value, the more biologically relevant it likely is.
Scroll even further down to find the “alignments” section, which provides the most detail available (Figure 3).
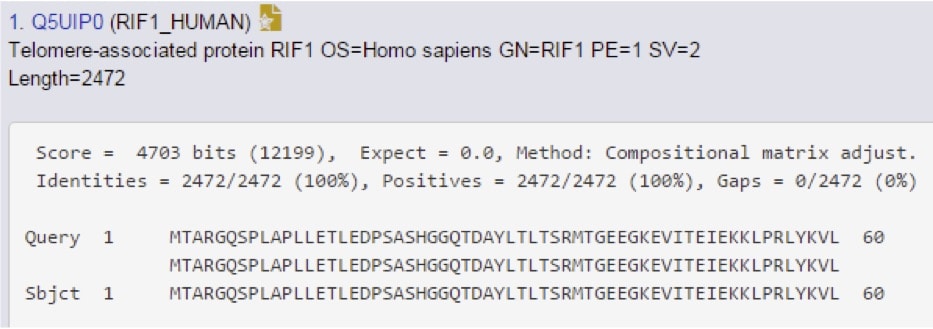
The score and E-value are re-stated here, but now you can see new information. This includes the “identities” section, which means “these two amino acids identical,” the “positives,” which can be read as “these two amino acids are different, but have similar chemistry,” and then gaps, which reflects any regions that are missing between the query and the subject sequences. In this case, the protein in question is human RIF1.
Is this Protein Found in a Different Species?
Knowing what we know now, this question is easy to answer. In Figure 2, the species name is a five-letter code following the gene name (i.e. HUMAN). If you see anything other than “HUMAN” in this space, you’ve answered your question.
To address specifically which species it is you’re looking at, scroll down to the view provided in Figure 3. The species name is provided on the second line in the “OS=” field.
Is this Protein Found Specifically in Mice? Again, How Similar is it?
Using our RIF1 example again, take Figure 2 and scroll down further to see if “mouse” is one of our options.

Yep, there it is. Notice the lower score though, so it’s not a perfect match. Let’s go see how imperfect it is…(Figure 5)
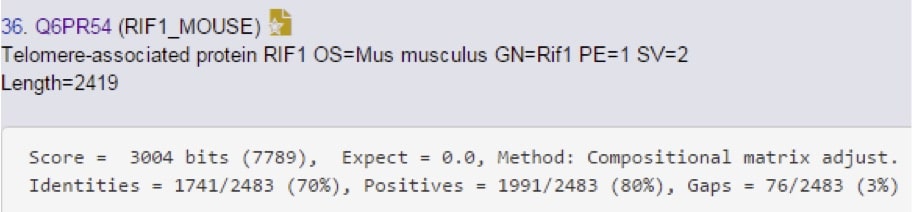
Showing the entire sequence would be silly, so we just take a look at the statistics here to see how similar it is. 70% identity is a pretty high degree of homology, and that increases to 80% if you consider side-chain chemistry similarity. These two proteins are highly homologous, but not identical.
Can I Find a Region of Sequence Novelty Between my Protein and a Group of Similar Proteins?
BLAST is a great tool for comparing one polypeptide chain to another, but it becomes unwieldy when you want to consider more than 2 sequences. In this case, use ClustalW, the most recent version of which is called Clustal Omega.
For the purposes of this example, I am using the first 120 amino acids of murine RIF1, with some small changes to illustrate how the program works. “Isoform 2” contains a series of point mutations in the first 10 residues, and “isoform 3” contains a 5 residue deletion from the C-terminus (Figure 6). Also, it is important to notice a small difference here when compared to BLAST. When you enter the sequences to align, you need to add a “>” followed by a title for your sequences. The program won’t run without it, and it doesn’t tell you this anywhere (Figure 6).

Upon clicking “submit,” the program simply lines the three sequences up, giving you an opportunity to quickly identify where the sequences are different. Take a look (Figure 7):

Here, you can very clearly see that isoform 3 and the wild-type sequence are conserved in the N-terminus. You also see that isoform 2 has the mutations. The program also suggests similarity, much like BLAST does. Serine (S) and threonine (T) have similar side chain chemistry, indicated by a “:”. Glycine (G) and lysine (K) do not, hence the blank. Further, isoform 2 and the wild type have similar C-termini. The gap present in isoform 3 is indicated by a “-“ in the sequence, allowing the alignment to stay oriented properly, but still emphasizing the lack of wild-type residue.
These examples only scratch the surface of what these two programs are capable of, but hopefully they provide a nice “on ramp” from which you can start exploring and finding creative ways to solve the actual problems you come across in your work.







