A Brief History of Detecting Amplicons
The Old Days of Ethidium Bromide
In the early 1990s, quantitative (q) PCR was in its infancy, and despite PCR itself already being around for 10 years, there were no easy ways of precisely quantifying the amount of DNA that was amplified in a PCR reaction. In those days PCR was expensive, labor intensive, and was fraught with technical issues such as carry-over contamination of PCR products, which made discerning the amount of amplified product rather tricky.
A team of scientists from Roche were among the first to come up with solutions to these problems through the use of a well-known fluorescent DNA binding dye – ethidium bromide. Addition of ethidium bromide to the PCR reaction during cycling meant that afterwards the samples could be placed under a UV light to detect endpoint fluorescent signal to determine the presence/absence of the desired target.
But this still left scientists with the problem of how to easily quantify the amount of product that was amplified and, therefore, how much DNA they started with. The qPCR revolution really began with the use of a camera to detect the generation of fluorescence in ‘real-time’ as the PCR cycling was progressing. The theory worked on the simple basis that a relative amount of DNA (PCR product) could be measured when the amount of reporter signal reached a certain threshold – the sooner that threshold was met, the more DNA was initially present in the sample. This continuous fluorescent measurement was the foundation for the qPCR technologies of today.
The Dawn of New Intercalating Dyes
Concerns over the safety and sensitivity of ethidium bromide led to the use of alternative fluorescent intercalating dyes, including SYBR® Green Dye, in the qPCR reaction. SYBR Green preferentially binds to double-stranded DNA, but with much greater affinity than ethidium bromide. SYBR Green and other intercalating dyes improved the sensitivity of detection meaning fewer PCR cycles were required. These dyes were also far more quantitative, as the emission of fluorescence was directly proportional to the amount of PCR product generated. Over twenty years later, intercalating dyes as a detection format still remains one of the most popular choices in basic science research.
qPCR Probes: Next Generation qPCR
So now we had a way to easily detect PCR product using fluorescent dyes and a much more sensitive method of quantification, but there were still some nagging problems to do with specificity of the amplified product and the uncertainty that the signal was attributed to the desired target. Remember that DNA-binding dyes bind to all double-stranded DNA (dsDNA). So it was impossible to distinguish between multiple products generated by non-specific primer annealing or spurious products from primer dimers.
A major development was the introduction of a third oligonucleotide sequence in the PCR reaction in addition to the forward and reverse primers. This extra sequence, known as the probe, replaces the need for DNA binding dyes. The probe is kitted out with fluorescent modifications that perform the business of generating fluorescent signal. The probe is designed to anneal specifically to the target PCR product generated during amplification and when hybridized, the probe emits signal in response to excitation at a particular wavelength, giving a proportional read-out of DNA quantity. Through the added security of the probe determining signal release, specificity of signal is improved compared to DNA binding-dye methods.
So just how do these qPCR probes work?
Dual-Labeled FRET Probes
The most common probe format is the dual-labeled probe, containing a reporter fluorophore that emits signal at the 5’ end and a 3’ quencher to extinguish that reporter fluorescence when not bound to the target. The fluorescent reporter dye becomes excited at a specific wavelength and emits this energy in the form of light at a longer wavelength. However, when the excited reporter dye is in close proximity to a quencher dye, which has an overlapping absorption spectrum, this light energy is transferred to the quencher. The light energy is then dissipated at the emission of the quencher, allowing the fluorophore to return to the ground state without releasing photons. This conversion of energy to the quencher is known as fluorescent resonance energy transfer (FRET).
Before binding to the DNA the fluorophore and the quencher can flop around at either end of the probe and come close enough to each other that the reporter dye is efficiently quenched. If the quencher is too far away for efficient quenching, then light energy is emitted. In qPCR, this occurs once the probe has bound to DNA to form a more rigid linear duplexed structure, and signals the generation of the PCR product.
Signaling Mechanisms
There are typically two signaling mechanisms acting during each round of PCR amplification that lead to the emission of fluorescent light from the separation of the fluorophore and quencher to promote emission of fluorescent light:
1. Hybridization to target – This occurs when the probe binds the intended DNA target. Annealing of the probe to the target strand linearizes the probe and increases the distance between fluorophore and quencher. The fluorescent reporter signal can now be emitted because the distance between these two dye molecules on the probe, the reporter dye and the quencher dye, is large enough to prevent efficient quenching.
2. Cleavage by polymerase – After hybridization, the fluorescence light signal from the probe is further enhanced when Taq polymerase cleaves the fluorophore off the probe using its 5’-3’ exonuclease activity. The hydrolysis event permanently separates the fluorophore from the quencher so the two entities are no longer linked. If the polymerase does not cleave the probe, it is simply displaced during extension and can re-enter its quenched state ready to participate in the next cycles. These two mechanisms operate together to create a robust signal during the reaction.
Hybridization and Cleavage mechanisms are illustrated in Figure 1 by observing fluorescence at both the denaturation and annealing temperatures. Hybridization results in a change in the levels of fluorescence between two points – signal is highest when the probe is bound, because quenching is prevented due to the linearization of the probe and is lower after it is melted off during denaturation where it again goes into its unbound quenched conformation. The remaining fluorescence after denaturation is due to signal from cleaved probe. Cleavage does not result in the same zig-zag pattern present during hybridization, given that once the dye and quencher are cleaved the reporter dye is left permanently unquenched. Instead the total level of fluorescence increases in proportion to the amount of probe cleaved.
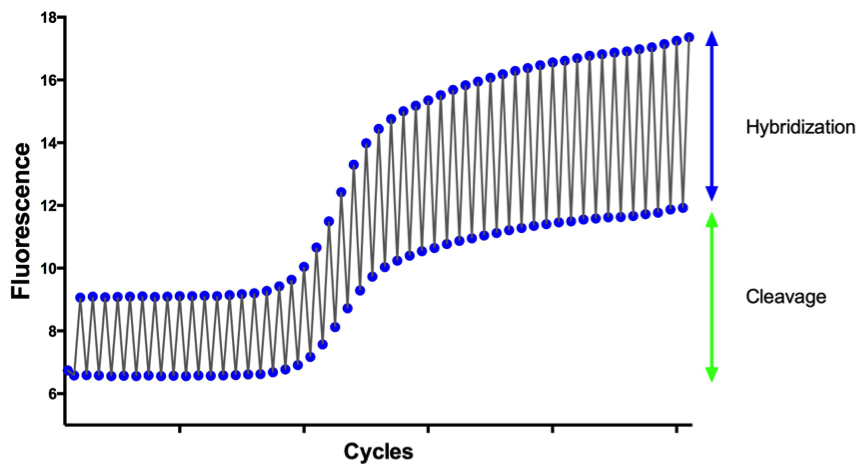
Figure 1. Visual representation of both Hybridization and Cleavage occurring during a PCR reaction.
What Can we do to Optimize Our Probe?
You can improve the chance of your probe providing a robust signal by ensuring that the melting temperature (Tm) of your probe is not too low – a low Tm will result in inefficient hybridization. Therefore, carefully designing the oligos is critical to assay performance. The probe should have a sufficiently high Tm, ideally a Tm above that of the primers, to allow it to bind strongly enough during the reaction to efficiently report signal.
Quenchers Snuff Out Dyes
Fluorescence resonance energy transfer is the physics behind light emission by dual-labeled qPCR probes. The choice of quencher–fluorophore pairing is critical to ensure that the fluorophore is adequately quenched. To achieve this, the emission spectra of the reporter fluorophore must adequately overlap with the absorption spectra of the quencher dye.
The Evolution of the Quencher:
TAMRA
The FAM-TAMRA pairing is a popular choice in FRET-based qPCR and was the first dye and quencher set to come about. When FAM (fluorophore) is excited by a specific wavelength, the energy is absorbed by TAMRA (quencher), which shifts the emission to the longer wavelength acceptor. Because TAMRA absorbs the energy from FAM and releases light at a longer wavelength, the fluorescent signal from FAM is effectively “quenched”.
While this was a popular option, this dye–quencher pairing does have its drawbacks, namely that TAMRA is fluorescent itself and, therefore, only quenches fluorophores with shorter emission wavelengths in the green and orange range. This severely limits the fluorophores that can be efficiently quenched by this dye, reducing the ability to multiplex. In addition, the TAMRA fluorescence can contribute to background signal leaking into the reporter signal and make qPCR measurements less precise.
The issues with lack of multiplexing capabilities and background noise were circumvented by the development of new quencher dyes, which did not have emissions in the visible spectrum, known as Dark Quenchers.
Dark Quenchers
The first dark quenchers that were developed included DABCYL, which unlike fluorescent quenchers (i.e. TAMRA), absorb excitation energy from the reporter dye (fluorophore) and dissipate it as heat rather than light. This property cuts out much of the problem of background signaling. Although DABCYL very efficiently absorbs light in the blue and green spectra, it can only absorb from a small range of wavelengths, which limits its use in multiplex qPCR applications.
BHQ® (Black Hole Quencher®) Dyes
The development of Black Hole Quencher (BHQ) dyes has been fundamental in the optimization of multiplex qPCR. BHQ dyes are a type of dark quencher that, like DABCYL, absorb light energy and re-emit it as heat, but do so at longer wavelengths as well. This opens them up to efficient quenching capability across the visible spectrum and allows pairing with a much wider range of fluorophores.
A number of BHQ dyes are now available, each absorbing light over a different range of wavelengths. This opens them up to pairing with a much wider range of fluorophores, allowing the simultaneous use of different reporter dyes. Overall the line of BHQ dyes has a quenching range covering from 430 nm right up to 730 nm and beyond (Figure 2).

Figure 2. Absorption spectra for BHQ dyes. Image is similar to some of those generated by Biosearch’s Spectral Overlay Tool.
Biosearch Technologies offers a neat, free-to-use spectral overlay tool, which is extremely useful when designing a multiplex qPCR experiment. The absorption and emission spectrum of multiple reporter–BHQ pairs can be easily determined with this tool, providing optimal multiplex probe pairings specific to your instrument for your next experiment.
This has simply been a quick guide to fluorescent qPCR probes and quenchers. To learn more check out the references below and be sure to visit Biosearch’s qPCR Design Lab.
References:
- Higuchi et al. (1992) Simultaneous amplification and detection of specific DNA sequences. 10:413.
- Higuchi et al. (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology 11:1026
- Dragan et al. (2012) SYBR Green I: fluorescence properties and interaction with DNA. J Fluoresc 22:1189
- Schneeberger et al. (1995) Quantitative detection of reverse transcriptase-PCR products by means of a novel and sensitive DNA stain. PCR Methods Appl 4:234
- Holland et al. (1991) Detection of specific polymerase chain reaction product by utilizing the 5′—-3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci USA 88:7276
- Tyagi and Kramer (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 14:303.
- Okamura et al. (2000) Double-labeled donor probe can enhance the signal of fluorescence resonance energy transfer (FRET) in detection of nucleic acid hybridization. Nuc Acid Res 28:e107
- Chen et al. (1998) Fluorescent, sequence-selective peptide detection by synthetic small molecules. Science 279:851