
The completion of the Human Genome Project in 2003 ushered in a new era of rapid, affordable, and accurate genome analysis—called Next Generation Sequencing (NGS). NGS builds upon “first generation sequencing” technologies to yield accurate and cost-effective sequencing results.
Fred Sanger sequenced the first whole DNA genome, the virus phage X174, in 1977. In that same year, Sanger developed the future backbone of the genome era: DNA sequencing technology. His technique used the chain termination method. This is now seen as the “first-generation technology” of genome sequencing.
Since Sanger sequencing (or the chain termination method) is the first generation of sequencing technology, understanding it is greatly important. The Human Genome Project began with Sanger sequencing technology.
The Sanger Sequencing Method
The Sanger sequencing method relies on dideoxynucleotides (ddNTPs),a type of deoxynucleoside triphosphates (dNTPs), that lack a 3′ hydroxyl group and have a hydrogen atom instead . When these bases bind to the growing DNA sequence, they terminate replication as they cannot bind other bases.
Choose a free resource to help you move forward
EBOOK
Gene Editing 101
DOWNLOAD
Blood Collection Tube Chart
To perform Sanger Sequencing, you add your primers to a solution containing the genetic information to be sequenced, then divide up the solution into four PCR reactions. Each reaction contains a with dNTP mix with one of the four nucleotides substituted with a ddNTP (A, T, G, and C ddNTP groups).
At the end of the PCR, each of your four reactions will yield PCR products of various lengths because replication is randomly terminated. By running the samples on a gel with 4 lanes, you can piece together the sequence as each sequence has been replicated from the same original material. Here is an example where the ddNTPs are in bold and the dNTPs are not:
Your sequence is ATGCTCAG. Your four reactions give you:
- Reaction with ddATP: A, ATGCTCA, ATGCTCAG
- Reaction with ddGTP: ATG, ATGCTCAG
- Reaction with ddCTP: ATGC, ATGCTC, ATGCTCAG
- Reaction with ddTTP: AT, ATGCT, ATGCTCAG
All the reactions once run a gel would look something like this (Image by Olwen Reina):
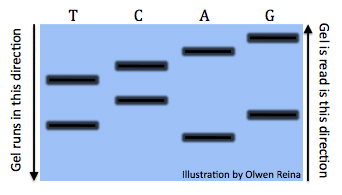
Each band denotes the different lengths code. For example, the band is the right under the “A” symbolizes the sequence: “ATGCTCA”
Still confused?
Let’s imagine a party game. The game is a guessing game. Here is how it is played:
You are thinking of a number and the group has to guess it. The tricky part is that the number is 200-digits in length. You are reading the digits of the number in your head without making a sound. Every so often a person interrupts you, and you tell them the single digit you were just thinking and where it is in the sequence of 200.
Each time you are interrupted, you have to start again. You leave after a few hours and the group has to figure out the 200-digit number. They have to piece together the information you gave them, for example the 25th number was 5, the 40th number was 0, and so on. Using the information from their interruptions, they can repeat the number they gave you.
While this sounds like the lamest game in the world, it works very well for sequencing!
Unfortunately, it is slow, expensive, and (previously) relies on radioactive materials. This pushed scientists to develop new and better forms of genome sequencing.
Next Generation Sequencing Technologies
The biggest advances in genome sequencing have been increasing speed and accuracy, resulting in a reduction in manpower and cost. These advances unlock striking applications, like NGS, which unearthed the fungus behind the Irish potato famine. Newer approaches push further still — see this CRISPR-inspired method that targets large, repetitive DNA elements.
The speed is thanks to parallel analysis and high-throughput technology.
The Sanger Sequencing Method
Sequencing machines have improved wildly since Sanger developed his method.
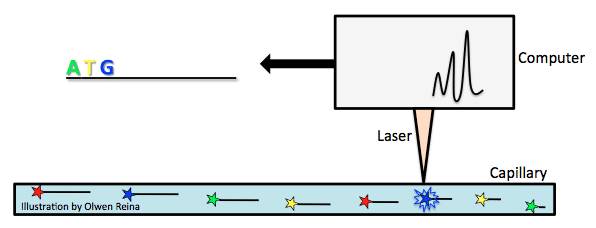
In 1987, Applied Biosystems became the first to introduce an automatic sequencing machine, called the AB370. The machine relied on a method called capillary electrophoresis that used Sanger’s chain-terminating method without needing a gel.
First, fluorescently labeled chain-terminating ddNTPs are added to the PCR reaction mix. By the Sanger sequencing method, PCR products of various lengths are created, and then separated according to their size. Size is measured by the PCR product’s overall negative charge. The more negative the charge, the longer the fragment.
Take the above example of ddNTP incorporation and imagine that the bold letters are ddNTPs with a fluorochrome attached. As the fragments are pulled toward the positive electrode of a capillary (see image below), they pass a laser beam that triggers a flash of light from the fluorochrome attached to the ddNTP that is characteristic of the base type (for example, green for A, yellow for T, blue for G, red for C). In this way, the genome is carefully read.
The biggest difference here was speed and cost. AB370 could detect 96 bases at one time, 500K bases per day, and had a read length of 600 bases by using a parallel analysis and high-throughput setup. This form of sequencing became the primary tool for completing the Human Genome Project.
How Does NGS Compare to First-Generation Sequencing?
NGS is characterized by improved accuracy and speed, but also reduced manpower and cost.
There has never been a time when it has been as cheap, convenient, or straightforward to sequence a genome. Arguably, the biggest improvement has been the development of parallel analysis, which has increased the sequencing speed.
The only thing slowing us down now is the interpretation of results! That interpretation increasingly touches new biology, like alternative polyadenylation.
Sometimes data interpretation gets complicated — here’s what to do when whole genome sequencing yields unexpected results. One free tool that helps is Galaxy, an NGS workflow management system. It also pays to know some Linux for high-throughput sequencing analysis and to be aware of batch effects, which can distort results across runs.
If you’re running many samples in parallel, it’s worth understanding probability theory and molecular barcodes. It’s also essential to quantify your NGS libraries accurately before pooling them and ensuring clean input before sequencing. For single-cell workflows specifically, see our tips to optimize nuclei extraction and counting.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.







