
As researchers, we are constantly on the lookout for new and improved ways to analyze, detect and quantify our favorite protein or gene. Luckily, we don’t always need to reinvent the wheel! PCR-ELISA is a good example of where two commonly used techniques have been merged together to create a very powerful analytical tool.
What is PCR-ELISA?
As the name suggests, PCR-ELISA combines an immunological method (ELISA) to detect and quantify specific PCR products directly after immobilization of DNA on a microtiter plate. PCR-ELISA is also less commonly known as PCR-ELOSA (polymerase chain reaction-enzyme-linked oligosorbent assay). This technique originated in the 1980’s and is most often used today in pathogen detection, but has broad applications across many fields (summarized in Box 1).
Box 1: APPLICATIONS FOR PCR-ELISA |
Diagnosis of virus in clinical samples (e.g. blood, serum, biopsies). |
Accurate diagnosis of pathogenic bacteria and fungi to species level ensuring appropriate treatment. |
Genotyping of individuals, for example, to identify carriers of recessive diseases. |
Environmental monitoring, for example, getting a clear picture on the exact nature of fungal and bacterial isolates in hospitals. |
Safety for example, rapid monitoring of salmonella in dairy products. |
Screening of blood donations to eliminate contamination in blood products (e.g. hepatitis B antigens, HIV-specific IgG). |
Epidemiology – to track the spread of pathogenic isolates and the appearance of new isolates. |
How does it differ to a regular ELISA?
Conventional ELISA is widely used to detect antigens of microbial, animal and plant origin, to detect circulating antibodies in serum (e.g. HIV-specific antibodies to diagnose and monitor infection), and in immunology to measure cytokine and chemokine levels to monitor autoimmune disorders and responses to treatments. Despite its many uses, the versatility and adaptability of ELISA is hampered by the requirement for purified monoclonal antibodies and antigens, which can be very time consuming to generate and require extensive optimization.
PCR-ELISA, on the other hand, is in principle capable of detecting genomic material from any biological specimen, as long as suitable primers can be designed to amplify the target DNA sequence. Although PCR-ELISA is unlikely to replace conventional ELISA or Real-Time PCR (see Real-Time PCR article) in the detection of proteins, biomolecules and nucleic acids, it does offer several advantages over these methods, and these are summarized in Box 2 at the end of this article.
Choose a free resource to help you move forward
DOWNLOAD
Blood Collection Tube Chart
POSTER
Top 10 Tips for Proper Pipette Care
How does PCR-ELISA work?
PCR–ELISA was initially developed to allow the direct analysis of biotin-labelled PCR amplicons using streptavidin-coated microtiter plates, but a hybridization step with specific probes was later incorporated in order to improve the sensitivity and specificity of the detection.
Nowadays, a sensitive and specific PCR–ELISA involves the labeling of target amplicons during PCR, their hybridization with target-specific probes, capture of labeled hybrids onto microtiter plates or tubes and subsequent detection by immunoassay. Labeling of amplicons is often performed using digoxigenin (DIG) molecules, and the DIG-labeled amplicons are easily detected using highly specific anti-DIG antibodies. The immunoassay itself is essentially carried out like a conventional ELISA.
PCR–ELISA assays are usually carried out in microtiter plates with colorimetric signal detection. Depending on the specific requirements, it is also possible to perform chemiluminescent detection with microplate luminometers or imaging systems, and other formats such as miniaturized plates and high-throughput assay methods are also a possibility. A hypothetical PCR-ELISA for the detection of bacterial isolates in food samples might proceed as follows: (illustrated in Figure 1 below).
1. Primer design
You typically design consensus primers to span a conserved genomic region within the bacterial genus/genera you are interested in, with enough variability to allow you to discriminate between species.
2. Probe design
For each target you want to detect, a unique and specific sequence of interest within the target sequence amplified using the consensus primer pair must be clearly defined as the target for probe design. Probes are generated by PCR using biotinylated primers.
3. PCR amplify target sequences
Target sequences are amplified from test samples. Depending on sample type, PCR may be applied on sample directly, or DNA purification may be necessary for complex samples. During the PCR reaction, the amplicons are labeled with digoxigenin (DIG) molecules so that they can later be detected with highly specific anti-DIG antibodies.
4. Hybridization of PCR amplicons with probes
PCR amplicons from test samples are incubated with with biotinylated probes under conditions that allow sequence-specific hybridization of DNA molecules.
5. Microtiter plate is coated with streptavidin
The plates are then coated with streptavidin which has a very strong affinity for biotin, and therefore binds to the biotinylated probes.
6. Immobilization of DIG-labelled PCR-probe hybrids
The hybrids are captured on the microtiter plate via streptavidin-biotin binding. The biotinylation of probes enhances specificity by ensuring that only amplicons bound to their specific probes are immobilized on this plate. This step helps to eliminate the immobilization of non-specific PCR amplicons and, therefore, reduces false positive results.
7. Addition of highly specific enzyme-labeled anti-DIG antibody conjugate
Anti-DIG antibody recognizes DIG-hybrids and remains in the plate. The antibody is usually conjugated to peroxidase or alkaline phosphatase.
8. Addition of colorimetric substrate
Substrate will undergo color change in the presence of conjugate, and this can be measured spectrophotometrically as for conventional ELISA.
Between each step, the microtiter plate is washed with a mild detergent solution to remove any unspecific hybrids or antibodies.
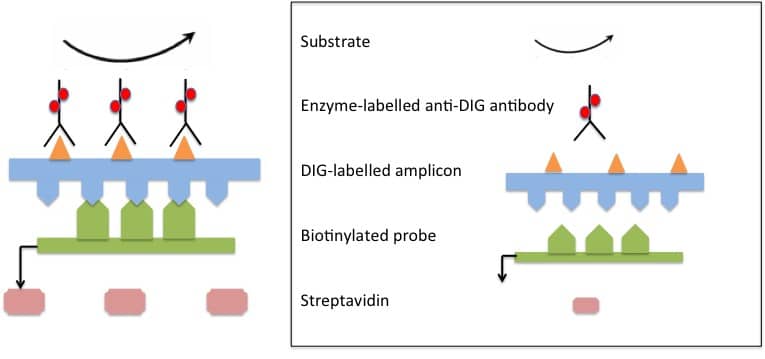
Figure 1: Scheme of PCR–ELISA. DIG-labelled target sequences (amplicons) are hybridized with specific biotinylated probes. Biotinylated DIG-labeled hybrids are immobilized onto a microtiter plate via biotin-streptavidin binding. Addition of specific anti-DIG antibody conjugate produces a colour change in the substrate, which is quantified spectrophotmetically. Figure adapted from Musiani et al., 2010.
PROTOCOL SUMMARY | CYTOKINES SECRETED |
1. Stimulate with immobilised anti-human CD3 antibody, soluble anti-human CD28 antibody, recombinant human IL-2 and IL-4. 2. Wash and culture with the same IL-2 and IL-4. 3. Harvest and restimulate with PMA, calcium ionophore or ionomycin. | IL-3, IL-4, IL-5, IL-13 and GM-CSF |
1. Stimulate with immobilised anti-human CD3 antibody and recombinant human IL-2. 2. Wash and culture in more IL-2. 3. Harvest and restimulate with PMA, calcium ionophore or ionomycin. Alternatively, restimulate with anti-CD3 and anti-CD28. | TNF- α |
Stimulate with PMA, calcium ionophore or ionomycin. | IL-2, TNF- α, and IFN-γ |
Stimulate with LPS. A 4-hour stimulation (a) will result in a different profile to a 24-hour stimulation (b). | (a) IL-1β, IL-6, IL-8 and GRO-α (b) IL-10, MCP-1, MIP-1α, MCP-3, and MIG |
Prime with recombinant human IFN-γ and LPS. | IL-12 |
Culture for 24 hours (as T cells constitutively produce this cytokine). | RANTES |
Important things to remember!!
- Positive and negative controls are critical. Controls are typically samples which have been proven unambiguously (through culturing, microscopy, pathological symptoms) to either contain your species of interest or not.
- Primer pairs should be optimized prior to use in PCR-ELISA set-up to find optimal annealing temperatures and rule out off-target amplification and primer secondary structures that might interfere with the assay.
- Samples are typically analyzed in triplicate to allow for pipetting errors, edge effects (a phenomenon of microtiter plates) and in-run variation.
At the end of the day, whether you choose to use conventional ELISA, Real-Time PCR or PCR-ELISA depends very much on your specific question or experiment. Conventional ELISA is very successful when the antigen or antibody of interest is well known, as kits are usually commercially available for well-known antigens, for example many human cytokines.
Despite the many advantages of PCR-ELISA as summarized in this article, it is not without limitations. It is difficult to match the accuracy in quantification offered by Real-Time PCR. Furthermore, the nature of PCR-ELISA restricts it to the detection of DNA and possibly RNA sequences, making it very useful in diagnosis of pathogens, but redundant in immunological research and the diagnosis of immune disorders and certain cancers (where specific proteins and peptides are measured).
Have you worked with PCR-ELISA? Have you found other applications for this technique besides what is described here? Please feel free to share your experiences with us.
Best of luck!
Box 2: Advantages of PCR-ELISA over conventional ELISA and/or Real-Time PCR |
Increased sensitivity. PCR-ELISA boasts increased sensitivity over conventional ELISA. By amplifying the target DNA sequence first, you increase the sensitivity of the assay to detect low abundant sequences. This may be very useful in the early detection and diagnosis of viral pathogens. |
Increased specificity. By detecting a species-specific DNA sequence, it is possible to confidently discriminate between different species, e.g. viral, bacterial or fungal pathogens. Conventional ELISA is often hampered by the fact that closely related species will have similar or the same antigens, leading to antibody cross reactivity and false positive results. |
Adaptable. Because amplified DNA is the target, commercially available species-specific PCR primers allow this technique to be established relatively quickly for different species. The same is true for Real-Time PCR. Conventional ELISA, on the other hand, requires a monoclonal antibody or a purified antigen (e.g. a viral envelope protein), which can be tedious, expensive and very time-consuming to generate. |
Faster throughput. Combining PCR with ELISA facilitates higher throughput since it omits the need for downstream DNA processing i.e. gel electrophoresis, purification and DNA sequencing. While a Real-Time PCR run itself (about 1 hour) is faster than an ELISA run, PCR-ELISA can still be time-saving since there is no requirement for nucleic acid purification from serum, cell and tissue samples. Depending on the type of microtiter plate used, 96 or 384 samples can typically be processed by PCR-ELISA in 6-7 hours. |
Automatable. PCR-ELISA can potentially be automated, due to robust standardization of the methods, reagents and samples. |
Cost-effective. PCR-ELISA can be performed with standard laboratory equipment, ruling out the need for expensive real-time PCR machines and DNA sequencing. |
Additional reading:
Musiani M, Venturoli S, Gallinella G, Zerbini M. (2007) Qualitative PCR–ELISA protocol for the detection and typing of viral genomes. Nature. 2:2502–10.
You made it to the end—nice work! If you’re the kind of scientist who likes figuring things out without wasting half a day on trial and error, you’ll love our newsletter. Get 3 quick reads a week, packed with hard-won lab wisdom. Join FREE here.







