Working with Enzymes: Part I -The Simple Kinetic Spectrophotometric Assay

At the end of my last article, I provided some practical tips and tricks for working with enzymes at the bench. Now, we’ll cover one of the cornerstone techniques of enzymology work: the enzyme assay.
Starting with the simple assays and eventually working our way to the more complex, this article introduces the principles of assaying enzymes spectrophotometrically. It relies on a friendly and relatively well-known piece of lab equipment used by biologists and chemists—the UV-Visible spectrophotometer (let’s just use the short-form “spec” for simplicity). The UV-Visible spectrophotometer does exactly what it says on the tin and detects light waves in the ultraviolet and visible spectrum.
However, you should know that there are other enzyme assays that will be covered later. For example, there’s binding assays that can be done using FRET or surface plasmon resonance (SPR), which the latter allows for label free detection. However, most protein laboratories have a spectrophotometer, and this assay is the most common. You must learn the basics before you can move onto other, more labor-intensive, techniques.
Assay Principle
A spec assay works by following a reaction as it progresses from substrates to products. For this to be meaningful, there must be a shift in the absorbance spectrum (or a spectral shift) of the biochemicals present that indicates that a change has taken place. By setting the spec to monitor a certain wavelength range, you can track the progress of the reaction by the disappearance of substrates or the appearance of products. Monitoring a reaction in real-time is a kinetic assay. An end-point assay is when you run the reaction for a pre-determined length of time but then treat the sample in some way before returning to the spec experiment.
As an example, I’m going to use something I studied from back in my own graduate days —glucose-6-phosphate dehydrogenase (G6PDH). In this reaction, glucose-6-phosphate (G6P) and oxidized nicotinamide adenine dinucleotide phosphate (NADP+) react to form 6-phosphoglucono-?-lactone (6PG?L) and reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADP+ absorbs negligibly at a wavelength of 340 nm, whereas NADPH absorbs strongly at this same wavelength (see diagram 1).
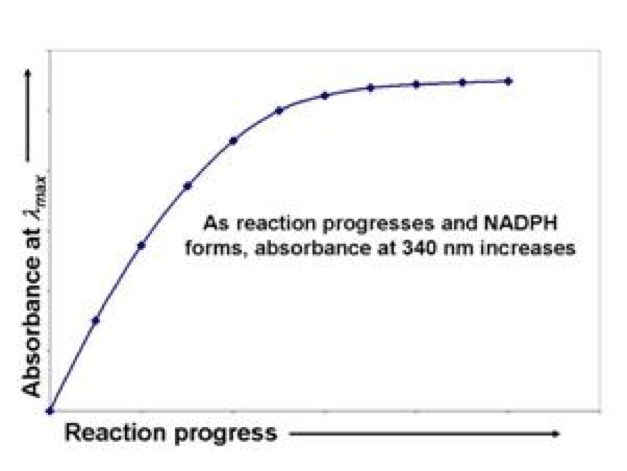
Diagram 1: The reaction: G6P + NADP+ ? 6PG?L + NADPH Tracking the appearance of NADPH at 340 nm
Assay components
For a simple spec enzyme assay, certain components need to be present:
- Chromophore/chromogenic substrate. The substrate is the chromophore or will become the chromophore after reacting. In my G6PDH study, the chromogenic substrate was NADP+, which reacted to form NADPH—the chromophore.
- Other substrate(s). If they are part of the reaction, other substrates need to be there. Otherwise, the biochemical reaction simply will not take place. With G6PDH, this was G6P. NADP+would not have reacted to form NADPH had G6P not been present.
- Cofactors, activators, ions. Ions or compounds that are not directly used up within the reaction but without them then the reaction will not proceed at an appreciable rate. Some Mg2+needed to be added to my G6PDH reaction in the form of MgCl2.
- Your enzyme of interest. This can be derived from animal tissue, plant extract, cell lysates, or any other source. The enzyme can be from crude isolation and full of contaminant proteins, partially purified, or purified to >98% homogeneity. Whatever the case, you will need to find the best method for obtaining your enzyme of interest, and what level of purity you need for your reaction.
- For consistency, you will want a final assay volume made of a pH-specific buffer, so that 1) there is enough “room” (volume) for all the assay components to be added at the highest expected concentrations of each, and 2) your enzyme is kept at the optimum pH and ionic strength for the reaction you are monitoring. For my G6PDH assay, this was always a 200 µL final volume in a 96-well microplate.
Notice that I have not even attempted to suggest any concentrations of substrates or other assay components, or the assay pH. This is all part of your individual optimization process. Looking through the literature at previous studies involving your enzyme will give you a starting point, but from there it is a process of scientific trial and error—or optimization, as it is commonly referred to—to determine what conditions work best for you.
Temperature
Just as it is up to you to optimize your substrate concentrations, so too is it necessary for other aspects of your assay, such as temperature. Last time, I indicated the importance of keeping your enzyme on ice until you were ready to assay it. You probably won’t run your assay at ice-cold temperature unless such a temperature has physiological relevance for your organism (as was the case with my freeze-tolerant frogs). In most cases, higher temperatures are desirable. “Room temperature” may suffice, but if your lab is in a building where this fluctuates wildly due to a poor HVAC system, then even a precisely duplicated enzyme assay can have strikingly different activities due to that temperature variation. Thankfully, this is the 21st century, and almost all kinetic assays are performed on temperature-controlled specs and allow you to set the temperature to your desired degree—25 °C, 30 °C, 37 °C, or whatever your temperature of interest may be. However, in order to get consistent results, remember to equilibrate the bulk of the reaction volume—usually the buffer and other stable compounds—to the reaction temperature (I usually do this in the spec) before the last, critical component is added to start the reaction.
Another word on assay temperature: your temperature of interest may or may not be the same at which your host organism normally lives. For a human enzyme, the automatic assumption may be 37 °C, but bear in mind that the dynamics of an enzyme in vitro are far different than they are in a living cell at physiological conditions. Yes, at some point you may want, or need, to assay your enzyme at your organism’s living temperature for the purposes of physiological relevance but that may not be your primary goal.
Starting the Assay and Assessing How Much Enzyme You Need
The vessel in which the enzymatic reaction is mixed and monitored could be anything from the small well of a transparent 96-well microplate, to a 1 mL or 2 mL cuvette, depending on the equipment at your disposal. In this vessel, mix all the components except for one—the enzyme (or one of the substrates critical to the reaction). This will depend on any specialized kinetic measurements you might be interested in. But realize that upon addition of that final component, your enzyme reaction begins immediately! As such, you need to have a measure of speed and dexterity.
Whether you are using modern software or an old-fashioned chart recorder, begin recording immediately after you have mixed your vessel and placed it back into the spec. At this point, you will also need to learn another important lesson: the concentration of your enzyme matters! Here’s why…
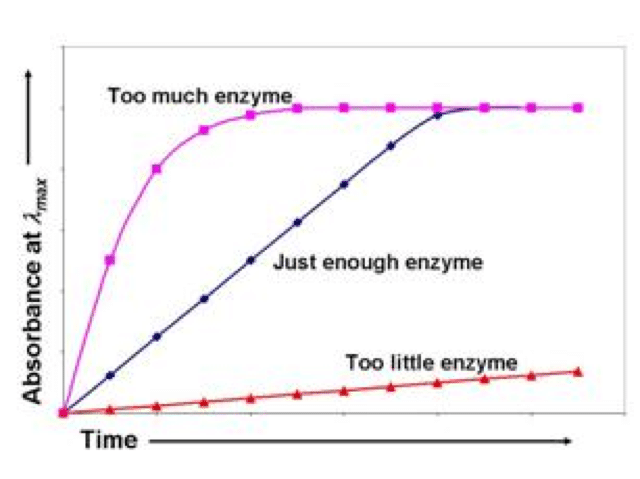
The more enzymes you have, the more binding sites of the enzyme are available to bind substrates. The more substrates that can be bound at any given time, the greater the turnover of your substrate into product. And so, high enzyme levels = high reaction rate.
The fewer enzymes you have, the fewer binding sites of those enzymes are available to bind substrates. Even if you cram all the substrate possible into your reaction vessel, less substrate can be bound at any given time because the few enzyme binding sites present will already be occupied by high levels of substrate. Less capacity to bind substrate leads to lower turnover of your substrate into product. And so, low enzyme levels = low reaction rate.
Overall, you want an enzyme assay where your reaction can be measured in an appreciable amount of time. If it progresses too quickly so that it is over by the time you mix your reaction vessel and begin recording, this is of no use to you. If it progresses too slowly so that it takes longer than a few minutes, this is also of no use to you- not to mention incredibly impractical (see diagram 2).
Beyond the Running of the Assay
What we have covered here are the most basic principles of making a simple spec enzyme assay work for you. Now that you have an assay up and running, how will you use it?
- Will you examine the basic kinetic parameters of your enzyme? How it binds its substrates and what affinity it has for them? What is the maximal rate of catalysis, (Vmax)?
- Will you vary your assay pH or temperature and determine the effect on your enzyme? The presence of increasing concentrations of ions and ionic strength? The effect of suspected activators and inhibitors?
- Will you throw denaturants like urea and guanidinium chloride at your enzyme and see how well (or poorly) its activity holds up against them?
Those decisions are up to you…
Next Time
It is very easy when your enzyme assay involves a reaction with a measurable spectral shift (G6PDH and NADPH). But what happens when your reaction of interest does not cause a spectral shift? In that case, the formation of a product must then be coupled to a secondary reaction (or more) which will produce the desired spectral shift. More to come!
Reference (my G6PDH study)
Dieni, C.A. and Storey, K.B. (2010) Regulation of glucose-6-phosphate dehydrogenase by reversible phosphorylation in liver of a freeze-tolerant frog. J. Comp. Physiol. B 180: 1133-1142.